
接種による重症化のリスクは否定できない!

ファイザー社が3月25日に提出した「コミナティ筋注 5~11 歳用に係る医薬品リスク管理計画書」には、重要な潜在的リスクとして疾患増強(ADE)が挙げられています。その理由として、「ADEによって重症化する可能性があると考えられる」と書いてありました。重要な不足情報として、「妊婦または授乳婦に接種した際の安全性」とあります。5歳から11歳への接種については、「製造販売承認時までに国内の 5~11 歳の小児での接種後の安全性情報はない」とはっきり書かれています。これらの情報は、医療現場で共有されているのでしょうか。
効果不良・薬効欠如の報告
厚労省のサイトで3月18日に公開された副反応疑いの報告を見ていると、これまでより「予防接種の効果不良」「薬効欠如」が増えていました。下記は、ほんの一部です。
ホーム> 政策について> 審議会・研究会等> 厚生科学審議会(予防接種・ワクチン分科会 副反応検討部会)> 第77回厚生科学審議会予防接種・ワクチン分科会副反応検討部会、令和3年度第30回薬事・食品衛生審議会薬事分科会医薬品等安全対策部会安全対策調査会(合同開催) 資料 3月18日
資料1-2-2-1より
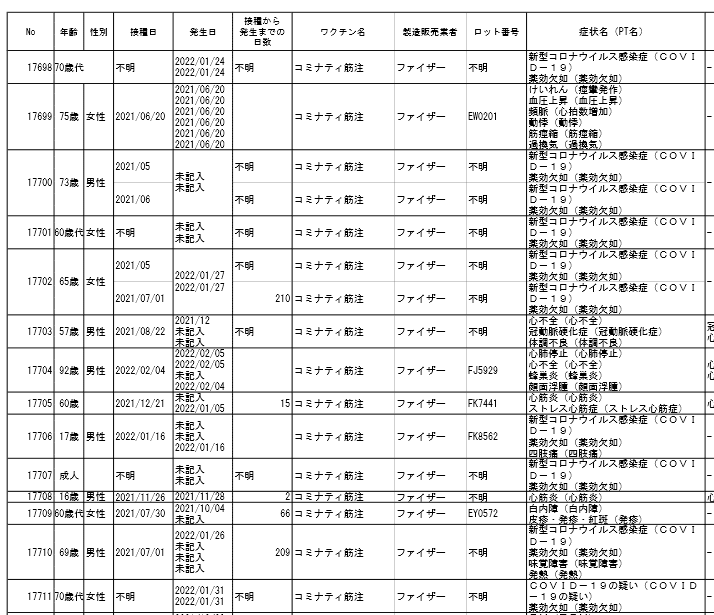
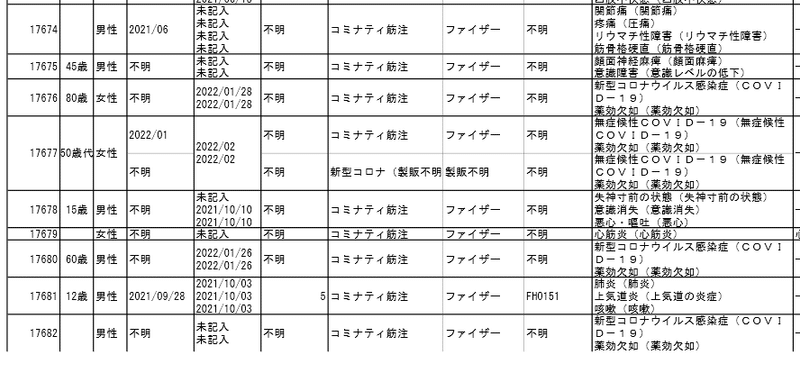

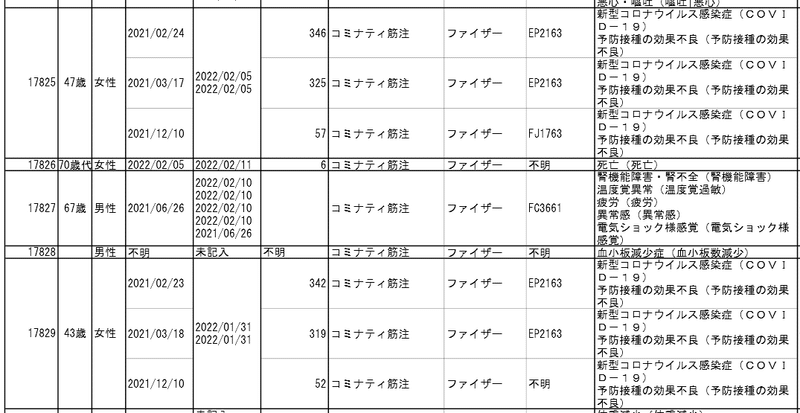
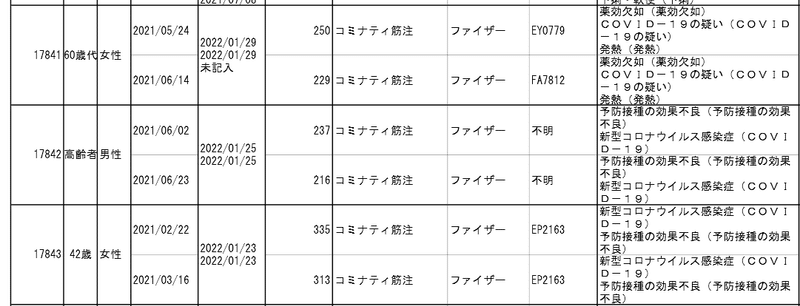

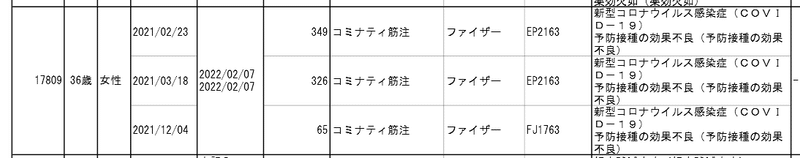
3回接種している事例について、資料1-2-3-1で詳しく見てみました。紹介する報告は、重複している部分などは編集してあります。時系列で追えるように、順番を入れ替えたりもしています。
事例:17809 36歳女性 コミナティ筋注3回接種
コミナティ筋注一般使用成績調査(承認後早期に接種される被接種者(医療従事者)を対象とした追跡調査)
本報告は、ファイザー社非介入試験(プロトコル番号:C4591006)からの報告である。
接種日:2021/02/23、ロット番号:EP2163、1回目
接種日:2021/03/18、ロット番号:EP2163、2回目
接種日:2021/12/04、ロット番号:FJ1763、3回目(ブースター)
関連する病歴:「同居家族が新型コロナウイルス感染症(COVID-19)となり、濃厚接触者に該当していた」(継続の有無は不明)。
併用薬は不明と報告された。
事前のワクチン接種後の有害事象は不明であった。
ワクチン接種日周辺の解熱剤使用は不明であった。
以下の情報が報告された:
2022/02/07、予防接種の効果不良(医学的に重要)、COVID-19(医学的に重要)が発現、転帰は「不明」。同居家族が新型コロナウイルス感染症(COVID-19)となり、被験者は濃厚接触者に該当していたと報告された。
2022/02/07(3回目のワクチン接種から2カ月3日後)、被験者は新型コロナウイルス感染症(COVID-19)を発症した。
2022/02/07(3回目のワクチン接種から2カ月3日後)、抗原定量検査実施し、陽性であった。事象が診療所来院もしくは緊急治療室来院を要したかは不明であった。
併用薬に対する事象の措置は該当なしであった。
調査担当医師は、事象を重篤(医学的に重要)と分類した。
調査担当医師は、重篤な有害事象と試験薬および併用薬との関連について合理的な可能性はないと考えた。
症状などは書かれていませんが、「事象を重篤(医学的に重要)と分類」としています。「C4591006」や「被験者」「試験薬」と書かれているのが気になりました。合理的な可能性とは、どのような場合に「あり」となるのでしょうか。
事例:17646 30歳女性 コミナティ筋注3回接種
コミナティ筋注一般使用成績調査(承認後早期に接種される被接種者(医療従事者)を対象とした追跡調査)
本報告は、プロトコル C4591006 のための非介入試験報告である。
2021/02/22(投与日)、ロット番号:EP2163、初回接種。
2021/03/15(投与日)、ロット番号:EP2163、2 回目接種。
2021/12/21(投与日)、BNT162b2(ロット番号:FK7441 3 回目(追加免疫)接種。
関連する病歴は以下を含んだ:
「COVID-19 陽性」、開始日:2022/01/26、終了日:2022/01/26。
併用薬は、報告されなかった。
2022/01/27、3 回目のワクチン接種の 37 日後に、被験者は COVID-19 陽性であった。事象の転帰は、未回復であった。
事象「COVID-19 陽性」は、医療機関の診察で評価された。
被験者は、救急救命室の受診を必要としなかった。
事象の経過は、以下の通りであった:
2022/01/26、被験者の職場の同僚が感染し、濃厚接触者となった為、PCR 検査対象となった。自覚症状がなかった。
2022/01/27、PCR 検査陽性が判明した。自宅療養となった。
調査担当医師は、以下の通りにコメントした:
現在自宅療養中の為、追加情報については分かり次第報告する。
解熱剤使用は、なかった。
調査担当医師は、事象「COVID-19 陽性」と BNT162b2 との因果関係について合理的な可能性がないと考えた。
こちらは、自覚症状がなかったケースです。症状がないのに「未回復」というのは、ずっと陽性だったということなのでしょうか。
なぜ今回、このような報告が多かったのかとても気になりました。 C4591006について調べていたら、ファイザー社が3月に提出した資料があり、それは非常に重要だと思うので取り上げます。
医薬品リスク管理計画
PMDAのサイトに、「コミナティ筋注 5~11 歳用に係る医薬品リスク管理計画書」(RMP)が公開されました。
医薬品リスク管理計画とは、下記のように説明があります。
医薬品の安全性の確保を図るためには、開発の段階から市販後に至るまで常にリスクを適正に管理する方策を検討することが重要です。医薬品リスク管理計画(以下、RMP)は、医薬品の開発から市販後まで一貫したリスク管理をひとつの文書に分かり易くまとめ、調査・試験やリスクを低減するための取り組みの進捗に合わせて、または、定期的に確実に評価が行われるようにするものです。また、RMPを公表して、医療関係者の皆様と市販後のリスク管理の内容を広く共有することで、市販後の安全対策の一層の充実強化が図られることが期待されます。
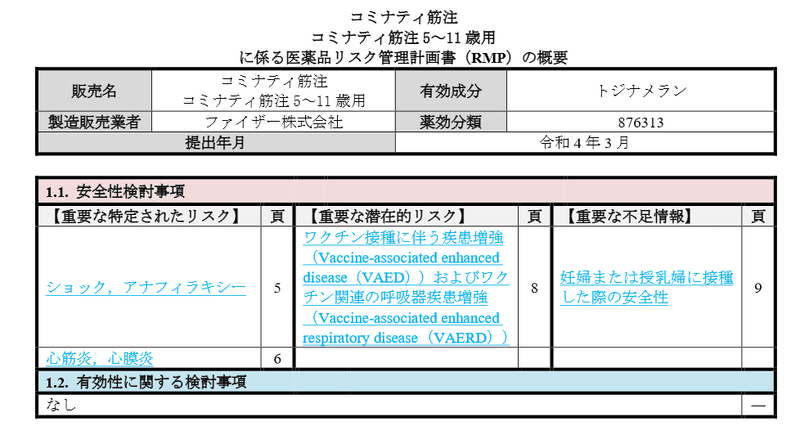
提出年月が、令和4年3月となっていますが、3ページに3月25日とありました。重要な潜在的リスクに「疾患増強」があり、重要な不足情報に「妊婦または授乳婦に接種した際の安全性」とあります。これは、とても重要なことではないでしょうか。
「3分でわかる!RMP講座」によると、潜在的リスクとは、「関連は疑わしいが十分に確認しきれないリスク」とのこと。臨床試験では十分に確認しきれていないので、可能性を否定できないということだと思います。
RMP講座のPDFには、「一歩進んだ薬剤師になるために!医薬品リスク管理計画書(RMP)の内容を理解し、活用しましょう!」と書いてあります。一歩進んだ薬剤師さんしか、RMPを活用しないのでしょうか。薬剤師さんは全員読んで、医師や看護師さんに伝えてほしいです。
ワクチン接種に伴う疾患増強(Vaccine-associated enhanced disease(VAED))およびワクチン関連の呼吸器疾患増強(Vaccine-associated enhanced respiratory disease(VAERD))
重要な潜在的リスクとした理由:
本剤の臨床試験において報告されていないものの,以下の報告を踏まえ,本剤の接種を受けた者が SARS-CoV-2 感染症に罹患した場合,VAED/VAERD により重症化する可能性があると考えられることから重要な潜在的リスクとした。
SARS-CoV-1 ワクチン候補を評価するために開発された動物モデル(マウス,フェレットおよび非ヒト霊長類)では,一部の研究で生ワクチン接種後のウイルス曝露時に疾患増強が認められた。
また一部の MARS ワクチン候補において,マウスモデルで疾患増強が認められた a) b)。
疾患増強の潜在的なメカニズムは,T 細胞媒介性(Th1 よりも Th2 による免疫病理学的反応)と抗体媒介性(中和活性が不十分な抗体反応が導く免疫複合体の形成および補体の活性化もしくはFc を介したウイルス侵入の増加)の両方であると考えられている c)。
特例承認に係る報告書(2021年2月)には、「現時点では不明」と書かれています。疾患増強については、当初から可能性を指摘する声が多くありました。特例承認に係る報告書では、「想定すると、リスクは低くなると考えられている」と書いてありますが、今回の計画書では「可能性があると考えられる」に変わっています。

ここで思い出されるのが、「ワクチンデマについて」(2021年6月24日)というタイトルで書かれたワクチン担当大臣(当時)が書いたブログの記事です。
「ADE(抗体依存性増強現象)が起きる」
ワクチンや過去の感染により作られる抗体が、ウイルスの感染を増強してしまうことをADEといいます。
デング熱ワクチンやSARSワクチンでこのようなことが起きたことがあります。
しかし、ファイザー社とモデルナ社のmRNAワクチンでは、高い中和作用がある抗体とバランスのよいリンパ球の動きが確認され、動物実験でもADEは観察されず、大規模な治験においてもADEの報告はないことから、新型コロナワクチンに関して、ADEの可能性は考えにくいとされています。
2021年6月24日に「ADEが起きる」というのをデマの1つとして挙げていましたが、ファイザー社が潜在的リスクとして挙げているなら、もうデマとは言えないでしょう。まだそのまま書かれていますが、訂正しないのでしょうか。
さらに、厚労省のQ&Aです。

今後も、新たな変異型が出現した場合には、ワクチンを接種した人でADEを含めた疾患増強が生じるかを観察する必要はありますが、現時点ではこのようなリスクの懸念はないと考えられます。
「現時点」というのがいつなのか、書かれていません。ファイザー社のリスク管理計画書が公開され、そこで重要な潜在的リスクと書かれたのだから、もう「懸念はない」とはいえないと思います。こちらもすぐに、書き換える必要があるのではないでしょうか。
承認後早期に接種される被接種者(医療従事者)を対象とした一般使用成績調査(追跡調査)(C4591006)

疾患増強などリスクに対する安全性監視のための活動に、「承認後早期に接種される被接種者(医療従事者)を対象とした一般使用成績調査(追
跡調査)(C4591006)」が挙げられています。これが、副反応疑いの報告に書かれていた「C4591006」です。
承認後早期に接種される被接種者(医療従事者)を対象とした一般使用成績調査(追跡調査)(C4591006)
【安全性検討事項】
・ワクチン接種に伴う疾患増強(VAED)およびワクチン関連の呼吸器疾患増強(VAERD)
・妊婦または授乳婦に接種した際の安全性
【目的】
本剤の製造販売承認後早期に接種される医療従事者(厚生労働省科学研究班が実施する先行接種者健康状況調査の参加者)を対象に,本剤による初回免疫の最終接種 28 日(先行接種者健康状況調査の観察期間終了日)経過後翌日から,本剤による初回免疫の最終接種 12 ヵ月後までの 11 ヵ月間追跡し,追跡期間中に認められた重篤な有害事象および COVID-19 情報を収集する。追加免疫を実施しなかった場合には,追跡期間中の本剤初回免疫後の長期的な安全性を確認する。追加免疫を実施した場合には,追加免疫の接種前日までの本剤初回免疫後の長期的な安全性を確認すると共に,追加免疫後も継続して重篤な有害事象および COVID-19 情報を収集する。
【実施計画】
調査期間:先行接種者健康状況調査の観察期間を最初に終了した症例の観察期間終了日翌日から本調査の最終調査対象者の観察期間終了まで(2021 年 3 月~2022 年 8 月を予定)
観察期間:本剤による初回免疫の最終接種 28 日(先行接種者健康状況調査の観察期間終了日)後翌日からの 11 ヵ月間
目標症例数:先行接種者健康状況調査の参加者(最大 20,000 例)のうち,本剤による初回免疫の最終接種 28 日後から本剤による初回免疫の最終接種 12 ヵ月後までの 11 ヵ月間の追跡調査への参加に同意が得られた登録条件を満たす被接種者全例
実施方法:EDC システムを用いる
主な調査項目:
・観察期間中に発現した重篤な有害事象および詳細情報
・COVID-19 病原体検査情報および COVID-19 情報
C4591006での安全性検討事項として、ワクチン接種に伴う疾患増強(VAED)およびワクチン関連の呼吸器疾患増強(VAERD)と、 妊婦または授乳婦に接種した際の安全性と書かれています。
ということは、3月18日にたくさん報告されているC4591006の事例は、疾患増強について確認するためなのでしょうか。
妊婦や授乳婦に関する情報不足については、下記のように書かれています。
本剤の生殖発生毒性試験において安全性上の懸念は認められていないものの,妊婦または授乳婦は承認前の臨床試験からはいずれも除外され,これまでの使用経験は少なく,妊婦または授乳婦に対する臨床上の安全性プロファイルは不明であるため,重要な不足情報とした。
不明というのに、なぜ医師たちは接種を勧めてきたのでしょうか。
このリスク管理計画書に書かれていることは、ワクチン推進に疑問を感じている人たちが以前から指摘していたことばかりです。
患者ではなく被験者
3月18日公開の副反応疑いの報告では、接種された医療従事者が「被験者」と書かれていることがとても気になりました。追跡調査への参加に同意が得られた登録条件を満たす被接種者が対象となっていますが、「被験者」となっているなら臨床試験に参加したのと同じような扱いなのではないでしょうか。
5歳から11歳への接種にも、同様のことが行われています。
5~11 歳の小児を対象とした特定使用成績調査(C4591032)
【安全性検討事項】
・ ショック,アナフィラキシー
・心筋炎,心膜炎
・ワクチン接種に伴う疾患増強(VAED)およびワクチン関連の呼吸器疾患増強(VAERD)
【目的】
使用実態下において,小児(5~11 歳)用製剤を初めて接種した 5~11 歳(接種日時点)の小児(基礎疾患を有する小児も含む)を対象に,小児(5~11 歳)用製剤接種後に認められる有害事象,局所反応,全身反応および COVID-19 情報を収集し,その安全性を早期に確認する。
【実施計画】
調査期間:最初の登録症例の 1 回目接種開始日から最終登録症例の観察期間終了日まで
目標症例数:検討中
観察期間:1 回目接種日から 2 回目接種後 28 日(約 7 週間)
(1 回目接種のみの場合は,1 回目接種後 28 日まで)
実施方法:EDC システムを用いた中央登録方式
主な調査項目:
・ 観察期間中に発現した全ての有害事象および詳細情報
・局所反応および全身反応[被接種者(保護者または法的代理人)が記録する健康観察日誌を使用]
・ COVID-19 病原体検査情報および COVID-19 情報
【実施計画の根拠】
小児を対象に実施した海外臨床試験(C4591007 試験)で得られた結果から,新たに追加された安全性検討事項はなかったが,製造販売承認時までに国内の 5~11 歳の小児での接種後の安全性情報はなく,製造販売後に安全性情報を収集する必要性は高いと考える。基礎疾患を有する小児も含め,国内の 5~11 歳の小児の安全性を確認する目的で特定使用成績調査を行う。
【目標症例数設定拠】
検討中
【節目となる予定の時期及びその根拠】
報告書作成時,安全性定期報告時および調査終了時に安全性の検討および報告を行う。
【当該医薬品安全性監視活動の結果に基づいて実施される可能性のある追加の措置及びその開始の決定基準】
本調査の結果を踏まえ,新たな安全性にかかわる懸念等が確認された場合または安全性検討事項に対するリスクが明らかになった場合,医薬品リスク管理計画書の見直しを行い,更なる検討が必要と判断する場合には,追加の医薬品安全性監視活動またはリスク最小化策の実施要否を検討する。
「基礎疾患を有する小児も含め,国内の 5~11 歳の小児の安全性を確認する目的」と書いてあります。つまり、国内での小児の安全性は、今、確認しているということです。そして、調査を進める中でリスクが明らかになる可能性があるということです。
テレビ番組では、昨日もADEのリスクには触れずに3回目の接種を勧めていました。テレビに出ている人たちは、この資料を読んでいないのでしょうか。読まずにコメントするのも問題ですが、読んでいても勧めるなら、なおさら信用できないと思います。