
肥満と糖尿病における炎症と代謝のバランスをとる微生物叢とNod様受容体

バイオメディカルジャーナル
2023年5月30日オンライン公開、100610号
In Press, Journal Pre-proofこれは何ですか?
レビュー記事
肥満と糖尿病における炎症と代謝のバランスをとる微生物叢とNod様受容体
https://www.sciencedirect.com/science/article/pii/S2319417023000471
著者リンク オーバーレイパネルRodrigo Rodrigues e-Lacerda, Han Fang, Nazli Robin, Arshpreet Bhatwa, Daniel M. Marko, Jonathan D. Schertzer
もっと見る
概要
シェア
引用元
https://doi.org/10.1016/j.bj.2023.100610Get 権利と内容
クリエイティブ・コモンズ・ライセンスに基づく
オープンアクセス
要旨
腸内細菌は、肥満時の宿主の免疫と代謝に影響を与える。自然免疫系の細菌センサーは、特定の細菌成分(すなわちポストバイオティクス)からのシグナルを中継し、宿主の代謝性炎症に相反する結果をもたらすことがある。Nod1やNod2などのNOD様受容体(NLR)は、ともに受容体相互作用プロテインキナーゼ2(RIPK2)をリクルートするが、血糖コントロールには相反する影響を与える。Nod1は細菌細胞壁由来のシグナルを代謝性炎症やインスリン抵抗性につなげるが、Nod2は免疫寛容、インスリン感受性、肥満時の血糖コントロールの改善を促進することができる。また、NLRP(NLR family pyrin domain containing)インフラマソームも、代謝に関する多様な結果をもたらす可能性があります。NLRP1はIL-18の制御に偏りがあるため、肥満や代謝性炎症から保護する可能性があるが、NLRP3はIL-1βを介した代謝性炎症とインスリン抵抗性に偏りがあるようである。免疫代謝を改善する特定のポストバイオティクスをターゲットにすることは、重要な目標である。Nod2リガンドであるムラミルジペプチド(MDP)は、肥満時や炎症性リポポリサッカライド(LPS)ストレス時の短時間作用型インスリン感作物質である。アンダーアシル化リピッドAを持つLPSはTLR4に拮抗し、炎症性LPSの代謝作用を打ち消す。Rhodobacter sphaeroides由来のアンダーアシル化LPSを与えると、肥満マウスのインスリン感受性が改善されました。したがって、ある種のLPSは、代謝的に有益な代謝性エンドトキシン血症を発生させることができる。保護的な適応免疫グロブリン免疫応答を関与させることで、肥満時の血糖値も改善することができる。上部腸の細菌群集全体の抽出物を用いた細菌ワクチンアプローチは、保護的な適応免疫応答と血糖コントロールの長期的な改善を促進します。今後の重要な目標は、血糖コントロールの改善に協力的なポストバイオティクスを特定し、組み合わせることである。


キーワード
T2D
肥満症
微生物叢
マイクロビオーム
NLRs
インスリン抵抗性
グルコース
インスリン
はじめに
肥満は、体脂肪蓄積の増加を特徴とする複雑な疾患である。肥満のリスクと進行には、食事、遺伝、座りがちなライフスタイル、環境など、多くの要因が関与しています。肥満は、2型糖尿病(T2D)を含む他の代謝性疾患のリスクを増加させます[1]。慢性的な低グレードの炎症は、肥満とT2Dに共通する特徴である。肥満による免疫系の変化は、脂肪組織、肝臓、骨格筋、膵臓、腸などの様々な代謝組織で起こります。肥満に関連した炎症は、表立った(細菌またはウイルス)感染時に比べて免疫反応の大きさが小さいという特徴がありますが、この代謝性炎症は慢性的でコンパートメント化することがあります[2]。白色脂肪組織(WAT)の炎症は、しばしば誘因となる事象であり、肥満の進行中に代謝における全身の変化を開始し、調整することができる [3]。
多くの異なるタイプの免疫細胞が、肥満時の代謝組織で増加する。例えば、マクロファージは、肥満の動物モデルと肥満の人の両方で肥大化したWATに浸潤しています[4]。マクロファージは、肥満時に増加する炎症性サイトカインの主要な供給源であり、マクロファージとその炎症性メディエーターは、組織および全身のインスリン抵抗性とT2Dの進行に関与しています[5,6]。腫瘍壊死因子α(TNF-α)はマクロファージが産生する炎症性サイトカインであり、インスリンの作用を変化させることができる。TNF-αの切除は、食事誘発性肥満マウスと遺伝子モデル肥満マウスの両方においてインスリン抵抗性を低下させる[7]。代謝細胞や免疫細胞内の多くの異なる免疫経路が、肥満時に活性化する。核因子κBキナーゼサブユニットβ(IKK-β)およびNF-κB経路の阻害は、肥満時のインスリン抵抗性を含む、炎症と代謝を橋渡しする。IKK-β/NF-κB依存性の炎症シグナルを薬理学的に阻害したり、遺伝的に欠失させることで、食事誘発性肥満におけるインスリン抵抗性の発症からマウスを保護することができます[8]。
肥満時の代謝性炎症の原因や誘因はよく分かっていませんが、いくつかの候補が同定されています。腸内細菌叢は、肥満時の炎症の起点となる可能性のあるものの一つです。Caniらは、高脂肪食(HFD)を短期間(すなわち4週間)摂取させると、代謝性内毒素血症として知られるリポポリサッカライド(LPS)の循環量が増加することを明らかにしています[9]。この画期的な研究は、グラム陰性菌の膜成分であるLPSが、肥満による炎症とインスリン抵抗性を引き起こす原因因子であることも示しています。LPSの上昇は、HFDによる腸内細菌叢の組成の変化と腸管透過性の増加の組み合わせの結果であると考えられる[10,11]。抗生物質、プロバイオティクスまたはプレバイオティクスの補充による腸内細菌叢の調節は、代謝性内毒素血症、炎症を低下させ、肥満時の血糖コントロールを改善し、腸内細菌叢が肥満関連炎症を変えるために標的となり得ることを実証している[11], [12], [13]]. 多くの研究が、微生物分類の変化と肥満時の代謝性炎症との相関を試みている。例えば、脂肪分が多く食物繊維が少ない食品は、腸内細菌叢の存在量と多様性を低下させ、Bacteroidetes/Firmicutes比を低下させます[14,15]。今後の重要な目標は、微生物叢の機能単位が免疫受容体とどのように関わり、代謝性炎症と宿主の代謝を変化させるかを明らかにすることである。
腸内細菌叢が宿主の免疫をプログラムし、影響を与えることはよく知られている[16]。肥満は、代謝性炎症と宿主の代謝に影響を与える宿主と微生物の関係を変化させる可能性があります。腸粘膜バリアと上皮は、微生物成分の宿主への移行を緩和する防御層である。脂肪組織や血液への細菌の移行レベルの増加は、1週間のHFD摂食ですぐに観察される[17]。HFDによる腸粘膜バリアーの崩壊は、上皮のタイトジャンクションタンパク質の減少を伴い、LPSの細胞外への移行を増加させ、代謝性内毒素症、炎症、代謝機能不全に寄与する [18].最近、肥満がエタノールアミン代謝の悪い微生物群を上腸管に発生させることが報告された。高濃度のエタノールアミンは、タイトジャンクションタンパク質であるzona occludens-1 mRNAと相互作用して不安定化させることができるmiRNA-101a-3pのプロモーター領域における転写因子ARID3の活性を高め、zona occludens-1 翻訳を減少させて、結果としてその発現を低下させることによって代謝性内毒素症や炎症の原因となる腸の透過性障害に関連していました [19].
LPSは自然免疫のtoll-like receptor(TLR)4に関与する。TLR4の発現と活性化は、T2D患者の単球で増加している[20]。TLR4の共受容体であるCD14の欠失によるLPS/TLR4シグナルの阻害は、LPSおよびHFD誘発の耐糖能と炎症からマウスを保護します[9]。これは、微生物叢、代謝性炎症、宿主の代謝をつなぐ自然免疫応答の一例に過ぎない。自然免疫系には、微生物叢由来のシグナルを中継して代謝の変化を引き起こすことができる細胞内コンポーネントが数多く存在する。ヌクレオチド結合オリゴマー化ドメイン様受容体、またはNOD様受容体(NLR)は、いくつかのインフラマソームを含み、宿主の代謝を変化させる微生物または危険なシグナルを検出します。例えば、NLRファミリー、ピリン・ドメイン含有タンパク質(NLRP)3インフラマソームは、自然免疫に関わる細胞質多タンパク質の「代謝危険センサー」であり、カスパーゼ-1の活性化を制御し、インターロイキン-1β(IL-1β)とインターロイキン-18(IL-18)の成熟とその後の放出をもたらす[21]。NLRP3やアダプタータンパク質ASC、カスパーゼ-1が存在しないマウスは、HFDによる肥満やそれに伴うインスリン抵抗性に抵抗性があり[22,23]、WATにおけるマクロファージの動員や炎症が少ない[24,25]。
本総説の目的は、肥満やT2Dの進行において、微生物が自然免疫や適応免疫に関わる細菌センサーとどのように関わり、代謝性炎症と宿主代謝に影響を与えるかを明らかにすることである。また、NOD様タンパク質が微生物叢と代謝性疾患をどのように結びつけているのか、そして、微生物叢からの様々なタイプのLPSが宿主の代謝にどのように影響を与えるのかについて言及する代謝性内毒素血症の概念を再考する。代謝を変化させる免疫応答で微生物を検出する
物理的な障壁に加え、自然免疫反応と適応免疫反応は、侵入してくる細菌からの保護をもたらす [26] 。防御準備の整った応答と免疫学的記憶の組み合わせは、宿主に対する多くの微生物の脅威を軽減することができる [27,28] 。しかし、宿主の代謝を変化させるには、自然免疫応答および/または適応免疫応答の関与が必要である。
自然免疫の一般的な概念は、病原体関連分子パターン(PAMPs)と損傷関連分子パターン(DAMPs)が、TLRやNLRを含む認識受容体(PRR)に関与することです。これらの免疫経路の共通ノードがNF-κBであり、IL-1β、IL-18、TNF-α、インターロイキン-6(IL-6)などの多くの炎症性サイトカインの転写が増加します[29,30]。NF-κBは、肥満時の代謝性炎症とインスリン抵抗性を媒介する可能性があります[31]。腸内細菌叢は、自然免疫反応のトリガーの源である [32]。常在菌、共生菌、病原性菌は、宿主に多くのユニークなPAMPsとDAMPsを提供します。LPS、ペプチドグリカン、フラジェリン、微生物の遺伝物質はすべて自然免疫の構成要素に関与し、宿主の代謝を変化させます [33] 。
適応免疫では、微生物由来の抗原が抗原提示細胞(APC)に関与し、エフェクターT細胞や制御性T細胞の機能を含むT細胞を指示することができる。微生物由来の因子は、Tヘルパー17(Th17)細胞にも影響を与え、その中には、分節した糸状菌からの大きな影響も含まれています[34]。Th17リンパ球は、IL-17ファミリーのインターロイキンの主な産生者である。IL-17EとIL-17Fの循環レベルはBMIの増加と正の相関があり、同じようにIL-17Eの循環レベルも皮下脂肪と正の相関がある。IL-17は、インスリン感受性を低下させることで脂肪組織にも作用し、長期的にはT2Dにつながる可能性があります[35]。食習慣もIL-17との相関を示し、野菜や果物の摂取は子どものIL-17を低下させるが、高度に加工された(ファースト)食品や飽和脂肪を多く含む食品の摂取はIL-17を上昇させる[36,37]。マウスのHFD後に、腸と脂肪組織でTh17サイトカインのレベルが低く、肝臓でこれらのサイトカインのレベルが高いことが観察され、肥満時のTh17免疫応答が区画化されていることが示された。腸におけるTh17反応の低下は、寛容な免疫環境と関連しており、代謝性内毒素血症や肝臓などの血糖コントロールに関わる代謝組織の炎症を引き起こす腸管粘膜バリアを越えた細菌成分の回避を促進する[38]。
重要な適応免疫反応のひとつが免疫グロブリンA(IgA)です。腸内細菌叢は、IgAの重要な影響因子であり、供給源でもあります[39]。腸由来の抗原は、M細胞と相互作用してIgAの合成を開始する。その後、腸管内腔のIgAは、粘膜保護をもたらし、代謝機能を制御することができる [40]。IgAは、慢性炎症および全身性炎症を低下させ、インスリン抵抗性および血糖異常の原因となる腸内細菌成分の侵入を防ぎます。IgAは、単球(マクロファージなど)からの炎症性サイトカインをダウンレギュレートする[41]。肥満マウスはIgA+免疫細胞が少なく、IgA+B細胞集団(特に腸内)の回復により、肥満マウスの血糖コントロールが改善されます[42]。脂肪の炎症とIgAの低下のサイクルを断ち切ることが、長期的な血糖コントロールを改善するメカニズムである。肥満や2型糖尿病時の代謝を変化させる細菌センサー
3.1. NLRC(NOD1およびNOD2)
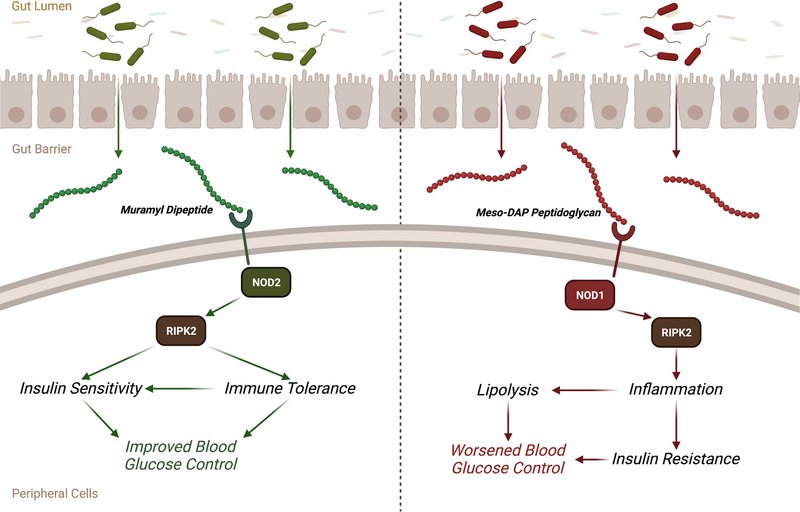
NLRは、大きくNLRCとNLRPの2つのサブファミリーに分けられる。NLRC は自然免疫の PRR であり、NOD1、NOD2、NLRC4、NLRC3、NLRC5、NLRX1 が含まれます。NLRCは、C末端ドメインにロイシンの繰り返し残基に富む領域(LRR)を持ち、中央にヌクレオチド結合オリゴマー化ドメイン(NOD)を持っています。また、N末端にはカスパーゼリクルートメントドメイン(CARD)があり、CARDの数や種類は様々で、CARDドメインを持たない場合もある[43]。NOD1および/またはNOD2は、インスリン抵抗性を含む代謝、および肥満時の血糖コントロールに影響を与えることができる[[44], [45], [46]] (図1)。NOD1とNOD2は、細菌細胞壁に由来するユニークなパターンのPAMPsに関与します。NOD1は主にグラム陰性菌と一部のグラム陽性菌のペプチドグリカンからγ-D-グルタミル-メソ-ジアミノピメリン酸(iE-DAP)を認識し、NOD2はグラム陽性菌に多く含まれるムラミルジペプチド(MDP)を認識できる [47]. 両分子の検出は、受容体相互作用プロテインキナーゼ2(RIPK2)のリクルートを誘導し、NF-κBとマイトジェン活性化プロテインキナーゼシグナルの活性化を頂点とする一連のイベントを開始する [48].
ダウンロード 高解像度画像ダウンロード(500KB)
ダウンロード フルサイズ画像のダウンロード
図1. NOD1シグナルとNOD2シグナルは、炎症と代謝に異なる影響を及ぼす。グラム陽性菌の細胞壁に含まれるムラミルジペプチドは、NOD2を活性化し、RIPK2に関与して、免疫寛容、インスリン感受性、血糖コントロールの改善につながる。一方、グラム陰性菌の細胞壁に含まれるメソDAPペプチドグリカンは、NOD1を活性化し、RIPK2に関与して、炎症の増加、インスリン抵抗性、血糖コントロールの悪化につながる。
NOD1とNOD2は、肥満によるインスリン抵抗性、代謝性炎症に関与していることが知られている[49,50]。マウスでNOD1とNOD2の両方を欠失させると、HFD食によって誘発される代謝性炎症とインスリン抵抗性が低下した [49].NOD1のみの欠失は、HFD食餌中の代謝性内毒素血症および生きた常在菌の腸関門を越えた脂肪組織への移行を低下させるのに十分であり、これは食餌誘導性インスリン抵抗性からマウスを保護する [17]. HFD誘発性肥満における細菌因子とインスリン抵抗性の相互作用は、造血器由来の免疫細胞のNOD1に関与している。免疫応答の中でも特に好中球が関与する代謝性炎症によって引き起こされる食事誘発性インスリン抵抗性からマウスを保護するには、NOD1の造血器欠失が十分であることが知られている[51]。食事誘発性肥満はまた、インスリン抵抗性と相関する循環中のNOD1リガンドを増加させ、NOD1リガンドは、代謝性内毒素血症と相乗して代謝性炎症と代謝性疾患を促進し得る別の微生物叢由来因子である[[51]、[52]、[53]]。NOD1リガンドを投与すると、マウスの脂肪組織と肝組織で全身のインスリン抵抗性と炎症が引き起こされた[49]。この研究は、NOD1が、特に脂肪組織においてインスリン抵抗性を促進する炎症反応に関与していることと一致する。NOD1の活性化は、脂肪細胞において過剰な脂肪分解、酸化ストレス、炎症、インスリン抵抗性を促進する[[54], [55], [56]]。実際、NOD1が介在する脂肪分解は、脂肪細胞における炎症反応を増幅させる[54]。さらに、メタボリックシンドロームや妊娠糖尿病のヒトでは、脂肪組織でNOD1レベルが高い[44,46]。したがって、NOD1を介した代謝性炎症がインスリン抵抗性を促進するという概念を支持する多くの証拠があり、NOD1は、微生物叢由来のシグナルを肥満時の宿主代謝異常に結びつける細菌センサーである[57]。
代謝性炎症の促進に加え、NOD1は正常な内分泌機能にも影響を及ぼす。例えば、膵臓β細胞におけるNOD1の活性化は、腸-膵臓軸を介してインスリン分泌を促進し、腸管リゾチームが腸内細菌叢から細菌細胞壁のムロペプチドを放出し、腸のバリアを通過して膵臓に作用する [58] 。NOD2リガンドは、インスリン分泌を変化させなかった[58]。したがって、特定の細菌のリガンドは、特定のNLRに関与して、正常な内分泌機能に影響を与えることができます。しかし、今後の重要な方向性は、肥満や代謝性疾患の特徴が、宿主の内分泌機能や代謝を変化させる細菌リガンドのバランスをどのように変えるかを明らかにすることです。肥満時の腸内細菌叢は、インスリンのクリアランスを変化させる単独要因である[59]。しかし、細菌のリガンドがどのように相互作用して多くの異なるホルモンを変化させるかについては、ほとんど知られていない。結局のところ、代謝の内分泌制御に対する微生物叢由来のリガンドの正味の効果は、NOD1とNOD2の相反する作用を含む、免疫刺激対寛容のバランスを表している。さらに、宿主と微生物の関係は双方向であり、血糖値などの宿主の代謝の特徴が、微生物叢内の共生、常在、寄生関係を変えることがある[60]。外的要因もまた、宿主と微生物の関係に影響を与え、免疫や代謝の結果に影響を与える可能性があります。食事性糖質は腸内病原体の増殖を促進し、分生子状糸状菌(SFB)を駆逐する。糖質によってSFBが減少すると、脂質の吸収を制御する腸の保護的な免疫反応が低下する。したがって、食事性糖質は腸管免疫を変化させ、脂質の過剰吸収を促進し、マウスの肥満を悪化させる可能性があります[61]。代謝疾患の免疫代謝の裏付けにおいて、外部からの合図や宿主と微生物の関係を考慮することは大変な作業であり、代謝の中枢と末梢の制御も考慮する必要がある[62]。
NOD2リガンドの急性投与は、筋肉細胞におけるインスリン抵抗性と筋肉組織における炎症を引き起こした[50,63]。しかし、NOD2のin vivoでの血糖コントロールとインスリン感受性に対する慢性的な作用は、NOD1と比較して非常に異なっている。NOD2の欠失は、HFD飼育マウスにおいてインスリン抵抗性を悪化させる[45]。NOD1とは対照的に、NOD2の造血器欠失はHFDによる血糖コントロールの変化に影響を及ぼさない。むしろ、非造血細胞におけるNOD2は、腸粘膜細菌のコロニー形成と代謝組織のディスバイオシスを制御し、代謝性炎症とインスリン抵抗性の増大の引き金となった[45]。代謝性疾患におけるNOD2のすべての作用に必要な細胞タイプは、まだ明らかになっていない。肝細胞内のNOD2は、腸-肝臓軸に関与することで、脂肪肝疾患の側面を制御することができる [64] 。しかし、肝細胞内のNOD2は、肥満マウスにおけるMDPのインスリン感作作用には必要ない [63]。重要なことは、NOD2リガンドをマウスに慢性的に投与することで、代謝性疾患モデルにおいて複数の利点があるということである。実際、NOD2リガンドであるMDPの投与は、体や脂肪組織量の変化とは無関係にインスリン感受性と血糖値を改善し、腸内細菌叢組成の変化も必要としない[63]。MDPは細菌由来のインスリン感作物質であり、NOD1が関与しない代謝性炎症の転写制御点であるインターフェロン調節因子4(IRF4)を必要とする[63]。全体として、異なる微生物叢由来の因子が宿主の代謝に及ぼす影響を媒介する上で、NOD1とNOD2が相反する役割を果たすという強い支持がある。今後の課題は、NOD1とNOD2が代謝性炎症と血糖値に対してどのように相反する役割を持つのか、特にこれらのNODタンパク質が共に共通のアダプターRIPK2を使用していることを考慮すると、それを明らかにすることです。今後は、RIPK2シグナル伝達の特定の側面が、NOD1とNOD2のリガンドが代謝に及ぼす影響の違いをどのように規定するのかを明らかにすることに焦点を当てるべきである。例えば、異なる細胞種における急性および慢性のNOD1およびNOD2活性化におけるRIPK2のユビキチン化状態は、免疫および代謝応答を生み出す可能性がある [65]. また、チロシンキナーゼ阻害剤がRIPK2の免疫代謝をどのように変化させるかについても評価する必要がある [66,67] 。また、代謝性疾患におけるNOD1、NOD2、IRF4の性差による影響を明らかにすることも重要である [68] 。
3.2. NLRP (NLRP1およびNLRP3)
NLRP1およびNLRP3インフラマソームは、免疫と代謝に影響を与えます(図2)。これらのインフラマソームは、多種多様なPAMPsやDAMPsを検出し、応答する[69]。NLRP1およびNLRP3インフラマソームの活性化は、脂肪組織や肝臓の代謝性炎症に関与しているIL-1βおよびIL-18の切断と活性化など、特定のサイトカイン応答を生成することができます[9]。一般に、これらのインフラマソームはプライミングと活性化のステップによって制御される。HFDによる腸内細菌叢の構成と腸管透過性の変化は、LPS/エンドトキシンなどの細菌成分のレベルを上昇させ、それによってNLRP3インフラマソームのプライミングシグナルを提供することができる[9]。二次(活性化)シグナルには、セラミド、飽和脂肪酸、活性酸素、壊死した脂肪細胞から放出されるATP、あるいはミトコンドリア機能障害によるものなど、多数の代謝的危険信号が関与する可能性がある [70].
ダウンロード: 高解像度画像ダウンロード(439KB)
ダウンロード フルサイズ画像のダウンロード
図2. NLRP1シグナルとNLRP3シグナルは、サイトカインと血糖コントロールに異なる効果を促進する。PAMPsとDAMPsは、NLRP1とNLRP3の両方を活性化し、IL-1βとIL-18を共に制御することができる。NLRP1を介したカスパーゼ-1の活性化は、プロIL-1 βとプロIL-18を切断するが、エネルギー消費を増加させ、肥満を抑制し、インスリン感受性を改善し、血糖コントロールを改善できる生物活性IL-18に偏りがあるようだ。一方、NLRP3-mediatd activation of caspase-1はpro IL-1βとIL-18の両方を切断するが、炎症、インスリン抵抗性、血糖コントロールの悪化を促進する生物活性IL-1βにバイアスがかかっているように思われる。
HFD摂食は、マウスの脂肪組織においてNLRP3とカスパーゼ-1の両方の発現を増加させる[22,23]。NLRP3の阻害または欠失は、食事や肥満に関連するインスリン抵抗性、血糖値異常、膵臓β細胞死を緩和することができ、これらはすべて、肥満によるインスリン抵抗性のT2Dへの進行の基礎となるプロセスである [69]. Pahwaらの研究によると、メタボリックシンドローム患者の皮下脂肪組織は、カスパーゼ-1、IL-18、IL-1βのレベルの上昇によって示されるNLRP3インフラマソーム活性が高いことが示されました。また、カスパーゼ-1の増加は、脂肪組織の線維化、血管新生、肥満細胞や好酸球の浸潤、インスリン抵抗性と関連していることが示された[71]。Antonioliらは、病的な肥満患者は、耐糖能とは無関係に、循環カスパーゼ-1が高いことを明らかにした。IL-1βの循環レベルは、正常な血糖値の肥満の人に比べて、肥満とT2Dの人で高かった。肥満手術後の減量は、循環カスパーゼ-1レベルを低下させたが、これは正常な血糖値の肥満の人にのみ観察され、肥満とT2Dの人には観察されなかった[72]。
脂肪組織の炎症は、肥満によって誘発されるインスリン抵抗性とT2Dに関与しており、脂肪細胞とマクロファージなどの組織常在の免疫細胞には、多くの冗長で重複した免疫応答が存在する[73]。肥満モデルや肥満者における代謝性炎症におけるNLRP3インフラソームの役割を示す圧倒的な証拠とは対照的に、Esserらは、NLRファミリーメンバーであるNLRP1が肥満者のWATで有意な増加を示さないことを実証した[74]。ヒトの肥満におけるNLRP1の役割を示唆するヒトでの確固としたデータはあまりありませんが、齧歯類の肥満モデルにはいくつかの証拠があります。複数の研究グループが、マウスでNLRP1を欠失させると、循環IL-18のレベルが低下し、脂肪率が上昇し、血糖コントロールが悪くなることを示している[75,76]。マウスにおけるNLRP1欠失の効果は、肥満の増加や異所性脂質の沈着など、IL-18欠失マウスの代謝表現型を反映しており、HFDまたは高タンパク質食によって悪化する影響である[75]。重要なことは,NLRP1が亢進し,IL-18が高いマウスは,食事誘発性肥満に抵抗性があることである.また、IL-18の過剰発現は、エネルギー消費を増加させることにより、HFDによって誘発されるインスリン抵抗性と血糖値異常からマウスを保護します[75]。
これまでのエビデンスから,肥満時の代謝性炎症におけるNLRP1およびNLRP3インフラムソームの役割は相反することが示唆される.これは、代謝性炎症と代謝性疾患リスクにおけるNOD1とNOD2の相反する役割に類似している。NLRP1は、IL-18の制御により、脂質の沈着から保護し、エネルギー消費を促進すると考えられる。NLRP3によるIL-1βの制御は、肥満時の代謝性炎症と代謝性疾患を示す組織機能不全を促進する。しかし、IL-1βとIL-18の両方を制御できるNLRP1とNLRP3が、どのように異なる代謝の結果をもたらすかは不明である。また、代謝性炎症において、NLRP1がIL-18に偏った反応を起こし、NLRP3がIL-1βに偏った反応を起こすかどうかも不明である。細胞種特異的なNLRP1対NLRP3応答は、異なる全身の代謝結果を生み出す重要な要因の一つである可能性がある。さらに、IL-1βとIL-18以外の各インフラマソームのカスパーゼ-1標的を明らかにし、固有のカスパーゼ-1切断標的の代謝的結果を明らかにすることは、今後の重要な目標です。例えば、NLRP3/カスパーゼ-1インフラマソームは、不活性なGAPDHを切断し、骨格筋の解糖能を低下させることができる [77]。
3.3. NLRsとTLRsの相互作用
TLRはパターン認識受容体であり、微生物成分を認識することで宿主の自然免疫応答の一部として機能します。TLRは、外来DNAやRNAを含む微生物、ウイルス、真菌由来のユニークなPAMPSのセットを認識し、LRRおよびトールインターロイキン1受容体(TIR)ドメインを介して免疫シグナル伝達カスケードを開始する [78,79] 。リガンドがLRR領域に結合した後に開始される特定のシグナル伝達経路は、TIRドメインに結合するアダプター分子によって決定される [79] 。主要なアダプターは、骨髄分化因子88 (MyD88) とTIRドメイン含有アダプター誘導IFN-β因子 (TRIF) である。下流のシグナル伝達は、IL-1β、IL-6、TNF-αなどの炎症性サイトカインの転写増加につながるNF-κBおよびインターフェロン制御因子の活性化をもたらすことができる[79]。TLRはよく研究され、特徴づけられ、レビューされています[79]。
肥満や糖尿病など、多くの慢性疾患状態がTLRシグナルの増加と関連している[9,80,81]。TLRは、IKKやJNKを含むストレスキナーゼの活性化につながるシグナルカスケードの活性化を通じて、インスリン抵抗性に影響を与える可能性があります [79]。これらのストレスキナーゼは、インスリン受容体基質タンパク質のセリン残基のリン酸化を引き起こし、インスリンシグナル伝播に必要なチロシンリン酸化を阻害する [78,82]. TLR4は、TLRが代謝性疾患に関与している重要な例である。TLR4はインスリン抵抗性を促進する[9,83,84]。CD14はLPS-TLR4シグナル伝達経路において重要であり、CD14変異マウスはHFDを摂取することでインスリン抵抗性が低下する[9]。肝細胞におけるTLR4の欠損は、マウスがHFDにより肥満になったにもかかわらず、耐糖能とインスリン感受性を改善する[85]。このように、TLRは代謝機能障害において主要な役割を担っていることが、証拠によって裏付けられています。
TLR と NLR は、相乗的な免疫応答や免疫寛容を生み出すために協力することができます。NOD1およびNOD2によるムロペプチドの認識とTLR4によるLPSの活性化を組み合わせると、IL-6、IL-1β、TNF-αなどの炎症性サイトカインが相乗的に増加します[86、87]。逆に、免疫寛容(すなわち、NLR活性化と組み合わせた場合のTLR活性化の抑制)もまた記録されている。慢性的または反復的な前処理は、NLRとTLRの間の免疫寛容を促進する重要な要因であるようです。Cavallariらは、野生型マウスにTLR4アゴニストである低レベルのLPSを急性注射する前に、3日間MDPを前投与し、代謝の評価を行った。MDP単独では血糖コントロールに影響を与えなかったが、LPSチャレンジの前にMDPを前投与すると、血漿インスリン濃度を変えずに血糖コントロールを改善した。重要なことは、MDPはNod2-/-マウスの耐糖能に影響を与えなかったことで、この相乗効果を媒介するためにNOD2が必要であることが示唆された[63]。NLRとTLRの間の免疫寛容は、リガンドと受容体に特異的である。NOD1活性化因子をあらかじめ注射し、その後LPSを急性注射すると、血糖コントロールが悪化することが、いくつかのグループによって確認されている [63,83]. TLRとNLRの異なる微生物トリガーに反応して、どの細胞タイプが相互作用して免疫寛容と相乗効果を促進するかはまだ明らかではありませんが、今後の研究では、NLRP1とNLRP3インフラマソームがどのように相互作用して宿主代謝を改善または低下させるかも検討すべきです。また、免疫相乗効果や寛容の観点から、肥満時に各組織がさらされる微生物リガンドレベルを理解することも重要であろう。さらに、血糖値、脂質、インスリンの上昇を含む肥満環境は、ある濃度のNLRおよびTLRリガンドにおける免疫相乗作用と耐性のバランスを変化させる可能性があります。NLRを介した代謝改善のための微生物叢の標的化
4.1. MDP
MDPはNOD2を介して肥満マウスのインスリン抵抗性を低下させるが、その際にIRF4が必要であった。NOD2に作用するMDPのインスリン感受性、血糖値低下作用にはIRF4が必要であったが、NOD1を活性化するムロペプチドが引き起こすインスリン抵抗性はIRF4が介在しなかった[63]。したがって、IRF4は細菌細胞壁の異なる部分によって引き起こされる血糖値応答を規定する転写因子であり免疫代謝のスイッチであるといえる。肥満時のMDPによるインスリン感作に関与する細胞型はまだ明らかではない。肝細胞ではNOD2を必要としないが、MDPは血糖コントロールを改善するために非造血のRIPK2を必要とする[65]。MDPの反復注射の主要な効果が脂肪組織の炎症を低下させることであることから、脂肪細胞特異的にIRF4を欠損させたマウスを試験したところ、脂肪細胞内のIRF4が性差に依存した形で血糖コントロールの改善に必要であることが判明しました。MDPは肥満の雄マウスでのみ血糖値を改善し、IRF4の脂肪細胞特異的欠失はMDPによる血糖値の改善を妨げた[68]。
MDPによる「忍容化」の期間は、MDPを3回(毎日)注射するだけでよく、食事誘発性肥満マウスや炎症型LPSに曝露したマウスにおいて、NOD2を介して脂肪組織の炎症とインスリン抵抗性を低下させた[63]。MDPによる寛容性を説明するために検証すべき重要な将来の方向性の一つは、免疫機能を規定することができる代謝の細胞再プログラミングである。IRF4は、好気性代謝を促進する代謝の「レオスタット」である。IRF4の欠失は、エフェクターT細胞における酸化的リン酸化と好気性解糖を損なう [88,89] 。脂肪組織マクロファージにおける好気性代謝の障害は、局所的・全身的な炎症と全身のインスリン抵抗性を高める。脂肪細胞における好気性代謝の障害は、炎症を増幅させる[90]。IRF4はT細胞受容体活性化時の酸化的リン酸化と好気性解糖を制御し、IRF4は肥満時の脂肪組織マクロファージにおける炎症を抑制することが知られている[89,91]。IRF4はまた、脂肪細胞における脂肪分解を制御している[91]。今後の重要な目標としては、初代脂肪細胞やマクロファージを含む脂肪組織常在の免疫細胞に対するMDPのテストが挙げられる。また、血糖値に対するMDPの性差を考慮すると、雌雄両方のマウスに由来する細胞を試験することも重要である。
4.2. 低アシル化LPS
肥満の原因である摂食によって炎症性LPSの循環レベルが上昇すると(すなわち、代謝性内毒素血症)、代謝性炎症と血糖異常につながる可能性がある [9,11,92]. 代謝性内毒素血症は、腸内細菌叢が慢性炎症、血糖値、インスリン抵抗性を変化させる作用機序として最も引用されるものの一つである[9、11]。代謝性内毒素血症は、TLR4アゴニズムを介して炎症と代謝異常を促進する大腸菌(E. coli)由来のLPSを用いて一般的にモデル化されてきた[9,11]。これまでの代謝性内毒素血症の概念では、LPSの構造における細菌株特有の変異を考慮していませんでした。ヘキサアシル化リピッドA(大腸菌)は高いTLR4活性化能を有するが、アンダーアシル化LPS(ペンタアシル化リピッドA)は用量依存的にTLR4と拮抗することができる[93]。代謝性内毒素血症の先行研究では、通常大腸菌に由来するヘキサアシル化リピドAを用いた炎症に焦点が当てられていた。しかし、最近のエビデンスでは、アンダーアシル化LPSを注射/投与することで、肥満マウスの血糖コントロール、血中インスリン低下、脂肪組織炎症が改善することが示されている[83]。したがって、アンダーアシル化LPSは代謝的に有益な内毒素血症を発生させると結論付けている[94]。
今後の課題としては、TLR4に拮抗することで代謝上のメリットを最大化する菌株の特定に注力すべきです。大腸菌は、TLR4の強力な活性化因子であるヘキサアシル化LPSを持つ傾向があり、ペンタアシル化LPSで操作した大腸菌は、TLR4拮抗作用だけでなく免疫原性の低下も示す [95]. 異なる菌種や菌株に由来するアンダーアシル化LPSは、程度の差こそあれ、TLR4の活性化に拮抗することができる[95,96]。
ロドバクター・スフェロイデス(R. sphaeroides)由来のペンタアシル化LPSは、大腸菌LPSによる腸管バリア機能、脂肪の炎症、インスリン生成能、血糖コントロールにおける劇症的変化に直接対抗できることが示された[94]。このことは、LPSの炎症性が代謝の変化に影響することを示唆している。また、R. sphaeroides LPSの慢性注入は、食事誘発性肥満が確立したマウスにおいてインスリン作用を増加させた[83]。今後の研究では、MDPや低アシル化LPSのような複数の有益なポストバイオティクスをどのように組み合わせるかを決定することにも焦点を当てるべきである。
4.3. 細菌ワクチン
腸関門を通過する特定の微生物叢由来因子は、肥満時の血糖値や宿主の代謝に逆の影響を与えることがある(図3)。すべてのポストバイオティクスが、血糖値やインスリンを含む宿主の代謝に及ぼす正味の影響は明らかでなかった。特定の消化管セグメント由来の腸内細菌エキスを特定用量注入することで、血糖コントロールが改善される可能性があった。投与量の最適化、腸の粘膜と内腔からのポストバイオティクス因子の抽出、超音波処理、滅菌の後、いくつかのグループは、上腸から希釈した細菌エキスの単回皮下注射が、痩せたマウスで1ヶ月後の血糖値の長期低下を引き起こすことを示している[97,98]。これは、肥満時の血糖値を改善することができる最初の細菌免疫化戦略の一つであった。無菌マウスから調製した粘膜抽出物はグルコースを低下させなかったので、この免疫化には細菌が必要だった [97,98]。細菌接種では、血糖値を下げるためにNOD2が必要であった。MDPだけではグルコース低下を再現することはできなかった[97]。したがって、細菌ワクチンによる血糖低下には、MDP-NOD2が必要であるが、十分ではない。したがって、内因性MDPは、血糖コントロールの長期的な改善を促進するために、別の細菌因子と共働する。腸内細菌由来の血糖降下ワクチンにおいて、これらの他の生物活性化合物がどのようなものであるかは、まだ明らかではない。細菌エキスを用いたワクチンでは、血糖コントロールを改善するために適応免疫が必要です[98]。そのため、代謝防御的な適応免疫反応を引き起こす細菌成分が重要な候補となる。
ダウンロード 高解像度画像ダウンロード(574KB)
ダウンロード フルサイズ画像のダウンロード
図3. 微生物叢由来のポストバイオティクスは免疫系を寛容化し、短期的および長期的な血糖コントロールの改善を促進する。ムラミルジペプチドはNOD2受容体に結合し、IRF4を必要とするため、免疫系を許容し、血糖コントロールの短期的改善を促進する。特定の細菌株由来の低アシル化LPSは、TLR4シグナルに拮抗し、それによって炎症を抑え、血糖コントロールの短期的な改善を促進することができる。細菌ワクチンのアプローチで腸上部に由来する細菌エキスを注射すると、保護的なIgA/IgG免疫応答が始まり、血糖コントロールの長期的な改善を促進する。
フラジェリンは、宿主の代謝に長期的な影響を与えることができる候補の1つです。フラジェリンは運動性細菌の鞭毛の成分であり、TLR5とNLRC4を通じて自然免疫を活性化する[99]。Tranらは、精製したサルモネラ菌由来のフラジェリンを繰り返し注射すると、血清および糞便中のフラジェリン特異的IgAおよびIgGレベルの上昇が引き起こされ、全身性の炎症が低下し、微生物叢の組成が変化することを示した[100]。IgAは、微生物叢が誘発する全身性炎症、インスリン抵抗性、血糖値異常を低下させるため、肥満時の代謝機能不全から保護する適応免疫応答である。腸管IgA+ B細胞を増やすと肥満マウスの血糖コントロールが改善されるので、IgAは肥満において重要である。細菌エキスやフラジェリン(MDPとの併用)によるワクチン接種が、腸内の保護的IgA応答を回復させるという推測は興味深い[42]。フラジェリンのワクチン的投与は、IL-10欠乏症誘発性大腸炎だけでなく、マウスの食事誘発性肥満からも保護する [100]. 肥満時の血糖値異常やインスリン抵抗性に対するフラジェリンワクチン戦略の効果を調査することが必要である。今後の重要な方向性は、血糖コントロールの短期的および長期的な改善を達成するために、候補となる生物活性化合物を組み合わせることであろう。また、微生物叢由来のポストバイオティクスに対する免疫寛容応答を通じて、どのNLRやその他の自然免疫応答が代謝に対する付加的な利益を伝播するのかを理解することも重要であろう。結論
NLRは、多くのDAMPSおよびPAMPsに対する免疫応答を伝播する。特定のNLRは、インスリン抵抗性を含む代謝性疾患の側面を促進する代謝組織における炎症性応答を中継することができます。肥満は、TLR4、NOD1、NLRP3を介する炎症性反応の活性化を促進する状況をもたらし、微生物叢由来の因子からの入力も含まれる。しかし、他の微生物因子を利用することで、これらの免疫代謝反応に対抗することができる。NOD2やNLRP1を介して免疫寛容を促進する微生物叢由来因子や、TLR4の活性化を阻害するLPS型は、代謝性炎症やインスリン感受性や血糖コントロールなどの宿主代謝を改善することができます。これらの短時間作用型ポストバイオティクスと、新しい細菌ワクチン戦略において保護的な適応免疫応答に関与するものを組み合わせることで、代謝性疾患の側面を改善する微生物叢由来因子と特定のNLR媒介免疫応答を標的とする方法を提供することができる。
競合する利害関係者の宣言
なし。
謝辞および資金源
本研究は、カナダ保健研究院(CIHR)の助成金FDN-154295からJDSへの助成金によって支援された。JDSは、代謝性炎症におけるカナダ研究チェアを保有している。DMはCIHR graduate scholarshipの支援を受けた。NRは、Natural Science and Engineering Research Councilの大学院奨学金により支援された。HFとABは、Farncombe Family Digestive Health Research Instituteのフェローシップと大学院奨学金により支援された。図はBiorender.comで作成した。
おすすめ記事
参考文献
[1]
S.E. Kahn、R.L. Hull、K.M. Utzschneider
肥満とインスリン抵抗性、2型糖尿病をつなぐメカニズム
ネイチャー, 444 (2006), pp.840-846
CrossRefView in ScopusGoogle Scholar
[2]
J.B.マクフィー、J.D.シャーツァー
肥満と糖尿病の免疫代謝:マイクロバイオータが腸内のコンパートメント化された免疫を代謝組織の炎症にリンクさせる
クリニカルサイエンス, 129 (2015), pp.1083-1096
スコープスで見るGoogle Scholar
[3]
J.M.オレフスキー
脂肪の話、肝臓と筋肉の話聞く
セル, 134 (2008), pp.914-916
PDFを見る記事を見るScopusGoogle Scholarで見る
[4]
S.P. Weisberg、D. McCann、M. Desai、M. Rosenbaum、R. L. Leibel、A. W. Ferrante
肥満は脂肪組織におけるマクロファージの蓄積と関連している
J Clin Invest, 112 (2003), pp.1796-1808
Scopusで見るGoogle Scholar
[5]
L. ハイルブロン、L.キャンベル
ヒト肥満における脂肪組織マクロファージ、低級炎症およびインスリン抵抗性
CPD, 14 (2008), pp.1225-1230
CrossRefView in ScopusGoogle Scholar
[6]
H. 神田
MCP-1は肥満症における脂肪組織へのマクロファージ浸潤、インスリン抵抗性および肝脂肪症に寄与する
ジャーナル・オブ・クリニカル・インベスティゲーション, 116 (2006), pp.1494-1505
CrossRefView in ScopusGoogle Scholar
[7]
K.T. Uysal, S.M. Wiesbrock, M.W. Marino, G.S. Hotamisligil
TNF-α機能欠損マウスにおける肥満誘発性インスリン抵抗性からの保護作用
ネイチャー, 389 (1997), pp.610-614
ScopusGoogle Scholarで見る
[8]
M.C. Arkan, A.L. Hevener, F.R. Greten, S. Maeda, Z.-W. Li, J.M. Long, et al.
IKK-βは炎症と肥満誘発性インスリン抵抗性を結びつける
Nat Med, 11 (2005), pp.191-198
CrossRefView in ScopusGoogle Scholar
[9]
P.D. Cani, J. Amar, M.A. Iglesias, M. Poggi, C. Knauf, D. Bastelica, et al.
代謝性内毒素血症は肥満とインスリン抵抗性を引き起こす
糖尿病, 56 (2007), pp.1761-1772
CrossRefView in ScopusGoogle Scholar
[10]
P.D. Cani, S. Possemiers, T. Van de Wiele, Y. Guiot, A. Everard, O. Rottier, et al.
腸内細菌叢の変化は、GLP-2による腸管透過性の改善を含むメカニズムを通じて、肥満マウスの炎症を制御する
Gut, 58 (2009), pp.1091-1103
CrossRefView in ScopusGoogle Scholar
[11]
P.D. Cani, R. Bibiloni, C. Knauf, A. Waget, A.M. Neyrinck, N.M. Delzenne, et al.
高脂肪食誘発性肥満および糖尿病マウスにおいて、腸内細菌叢の変化が代謝性内毒素血症誘発性炎症を制御する
糖尿病, 57 (2008), pp.1470-1481
CrossRefView in ScopusGoogle Scholar
[12]
M. Serino, E. Luche, S. Gres, A. Baylac, M. Bergé, C. Cenac, et al.
高脂肪食への代謝適応は、腸内細菌叢の変化と関連している
Gut, 61 (2012), pp.543-553
CrossRefView in ScopusGoogle Scholar
[13]
P.D. Cani, A.M. Neyrinck, F. Fava, C. Knauf, R.G. Burcelin, K.M. Tuohy, et al.
腸内細菌叢におけるビフィズス菌の選択的増加により、内毒素血症に関連したメカニズムでマウスの高脂肪食誘発糖尿病が改善される
Diabetologia, 50 (2007), pp.2374-2383
CrossRefView in ScopusGoogle Scholar
[14]
C. Zhang、M. Zhang、X. Pang、Y. Zhao、L. Wang、L. Zhao
高脂肪食摂食下における成体マウスの腸内細菌叢の構造的回復力
ISME J, 6 (2012), pp.1848-1857
CrossRefView in ScopusGoogle Scholar
[15]
R.E.レイ、P.J.ターンボー、S.クライン、J.I.ゴードン
肥満と関連するヒト腸内細菌
ネイチャー, 444 (2006), pp.1022-1023
CrossRefScopusGoogle Scholarで表示する。
[16]
H.-J. Wu、E. Wu
免疫恒常性維持と自己免疫における腸内細菌叢の役割
腸内細菌, 3 (2012), pp.4-14
CrossRefGoogle Scholar
[17]
J. Amar, C. Chabo, A. Waget, P. Klopp, C. Vachoux, L.G. Bermúdez-Humarán, et al.
2型糖尿病発症初期における常在菌の腸管粘膜付着と転位:分子機構とプロバイオティクス治療
EMBO Mol Med, 3 (2011), pp.559-572
CrossRefView in ScopusGoogle Scholar
[18]
M. Gulhane, L. Murray, R. Lourie, H. Tong, Y.H. Sheng, R. Wang, et al.
高脂肪食は大腸上皮細胞のストレスと炎症を誘発し、それはIL-22によって逆転される
サイレップ、6(2016)、記事28990
Scopusで見るGoogle Scholar
[19]
H. ファング、R.R.エ・ラセルダ、J.D.シャーツァー
肥満はエタノールアミン代謝する腸内細菌叢を低下させることでリーキーガット、炎症、糖尿病予備軍を促進する
ガット(2023)
GUTJNL-2023-329815
グーグルスカラー
[20]
M.R.ダス、S.デバラジ、S.パーク、I.ジアラル
最近診断された2型糖尿病患者におけるToll-Like Receptor(TLR)活性化およびTLRリガンドの増加
糖尿病ケア, 33 (2010), pp.861-868
CrossRefView in ScopusGoogle Scholar
[21]
K. シュローダー、R. Zhou、J. ツォップ
NLRP3インフラムソーム: メタボリックデンジャーを感知するセンサーか?
サイエンス, 327 (2010), pp.296-300
CrossRefView in ScopusGoogle Scholar
[22]
B. バンダンマグザー、Y.-H. Youm, A. Ravussin, J.E. Galgani, K. Stadler, R.L. Mynatt, et al.
NLRP3 インフラマソームは肥満による炎症とインスリン抵抗性を誘発する
Nat Med, 17 (2011), pp.179-188
CrossRefGoogle Scholar
[23]
R. Stienstra, J.A. van Diepen, C.J. Tack, MdH. Zaki, F.L. van de Veerdonk, D. Perera, et al.
インフラマソームは肥満とインスリン抵抗性の誘導における中心的なプレーヤーである
Proc Natl Acad Sci USA, 108 (2011), pp.15324-15329
CrossRefView in ScopusGoogle Scholar
[24]
H. Xu, G.T. Barnes, Q. Yang, G. Tan, D. Yang, C.J. Chou, et al.
脂肪における慢性炎症は、肥満に関連したインスリン抵抗性の発症に重要な役割を果たす
J Clin Invest, 112 (2003), pp.1821-1830
Scopusで見るGoogle Scholar
[25]
R. カンセロ、C. ヘネガー、N. ヴィグリー、S. タレブ、C. ポイトゥ、C. ルオー、et al.
手術による減量後の病的肥満者の白色脂肪組織におけるマクロファージ浸潤の減少および化学誘引性遺伝子発現の変化
糖尿病, 54 (2005), pp.2277-2286
CrossRefView in ScopusGoogle Scholar
[26]
D.D.チャップリン
免疫反応の概要
アレルギーと臨床免疫学ジャーナル, 125 (2010)
S3-23
グーグルスカラー
[27]
J.S.マーシャル、R.ウォリントン、W.ワトソン、H.L.キム
免疫学と免疫病理学への入門
アレルギー喘息臨床免疫学, 14 (2018), p.49
スコープスで見るGoogle Scholar
[28]
S.E.ターベイ、D.H.ブロイデ
イネイト免疫
アレルギーと臨床免疫学ジャーナル, 125 (2010), pp.s24-s32
Google Scholar
[29]
A.C.M.B. Gomes Torres, N. Leite, L.V. Tureck, R.L.R. de Souza, A.C.K. Titski, G.E. Milano-Gai, et al.
Toll様受容体(TLR)およびNOD様受容体(NLR)多型と脂質およびグルコース代謝の関連性
遺伝子, 685 (2019), pp.211-221
PDFを見る記事を見るScopusGoogle Scholarで見る
[30]
V.A.K. ラティナム、K.A. フィッツジェラルド
インフラマソーム複合体: 新たなメカニズムとエフェクター機能
セル, 165 (2016), pp.792-800
PDFを見る記事を見るScopusGoogle Scholarで見る
[31]
J.E.ベリザリオ、J.フェインチュ、M.ガレイ=マルパルティダ
腸内細菌叢の異常と免疫代謝: 代謝性疾患治療のための新たなフロンティア
炎症のメディエーター, 2018 (2018), pp.1-12
CrossRefGoogle Scholar
[32]
L.C.ランキン、D.アーティス
宿主防御を超える: 複雑な組織生理を制御する免疫系の新たな機能
セル, 173 (2018), pp.554-567
PDFを見る記事を見るScopusGoogle Scholarで見る
[33]
H. コノ、K.L.ロック
死にゆく細胞はどのように免疫系に危険を知らせるのか
Nat Rev Immunol, 8 (2008), pp.279-289
CrossRefScopusGoogle Scholarで表示する。
[34]
Q. ザオ、C.O.エルソン
腸内細菌叢抗原による適応的免疫教育
イムノロジー, 154 (2018), pp.28-37
CrossRefScopusGoogle Scholarで見る
[35]
E. ポラック・シュチビウォ、J・タバルキエヴィッチ
肥満者における低級炎症の強度と心血管疾患リスクの潜在的バイオマーカーとしてのIL-17A、IL-17EおよびIL-17F
ニュートリエンツ, 14 (2022), p.643
CrossRefScopusGoogle Scholarで表示する。
[36]
Y.-Y. ハン、E.フォルノ、J.M.ブレム、E.アコスタ=ペレス、M.アルバレス、A.コロン=セミディ、他。
プエルトリコ人における食事、インターロイキン-17、および小児喘息
アレルギー・喘息・免疫学年報, 115 (2015), pp.288-293
CrossRefGoogle Scholar
[37]
I.T.W. Harley, T.E. Stankiewicz, D.A. Giles, S. Softic, L.M. Flick, M. Cappelletti, et al.
IL-17シグナルはマウスの非アルコール性脂肪性肝疾患の進行を促進する
ヘパトロジー, 59 (2014), pp.1830-1839
CrossRefScopusGoogle Scholarで表示する。
[38]
J.F. Cavallari, E. Denou, K.P. Foley, W.I. Khan, J.D. Schertzer
肥満時の腸、肝臓、脂肪組織における異なるTh17免疫:食事、遺伝、微生物の役割
腸内微生物, 7 (2016), pp.82-89
CrossRefScopusGoogle Scholarで表示する。
[39]
G.L. Talham, H.-Q. ジアン、N.A.ボス、J.J.セブラ
セグメント化された糸状菌は、マウス腸粘膜免疫系の生理的に正常な状態の強力な刺激となる。
インフェクトイミュン, 67 (1999), pp.1992-2000
ScopusGoogle Scholarで見る
[40]
L. ドレ、H.Q.トラン、L.エチエンヌ=メスミン、B.シャッサン
適応免疫系による腸内細菌叢の取り締まり
BMC Med, 14 (2016), p. 27
ScopusGoogle Scholarで見る
[41]
H. ウルフ、M.フィッシャー、H.プーリンガー、A.サムスターク、E.フォーゲル、M.エイブール
ヒト血清IgAはヒト単球における炎症性サイトカイン(腫瘍壊死因子α、インターロイキン6)の放出を抑制する
血液, 83 (1994), pp.1278-1288
PDFを見る記事を見るScopusGoogle Scholarで見る
[42]
H. ラック、S.カーン、J.H.キム、J.K.コープランド、X.S.レベロ、S.ツァイ、他。
腸管関連IgA+免疫細胞は肥満に関連するインスリン抵抗性を制御する
ネイチャーコミュニケーションズ, 10 (2019), p.3650
スコープスで見るGoogle Scholar
[43]
M. ゴドコヴィッチ、M. ドルシチンスカ
抗ウイルスおよび抗マイコバクテリア免疫におけるNOD1、NOD2、およびNLRC5レセプター
ワクチン, 10 (2022), p. 1487
CrossRefView in ScopusGoogle Scholar
[44]
Y.-J. Zhou, C. Liu, C.-L. Li, Y.-L. Song, Y.-S. Tang, H. Zhou, et al.
メタボリックシンドローム患者の皮下脂肪組織におけるNOD1活性の上昇、ただしNOD2活性は上昇しない: NODとメタボリックシンドローム
肥満, 23 (2015), pp.1394-1400
CrossRefScopusGoogle Scholarで見る
[45]
E. Denou, K. Lolmède, L. Garidou, C. Pomie, C. Chabo, T.C. Lau, et al.
NOD 2ペプチドグリカンセンシングの欠陥は、食事誘発性の炎症、ディスバイオシス、インスリン抵抗性を促進する
EMBO Mol Med, 7 (2015), pp.259-274
CrossRefScopusGoogle Scholarで表示する。
[46]
M. ラパス
妊娠糖尿病の女性の脂肪組織でNOD1の発現が増加している
内分泌学雑誌, 222 (2014), pp.99-112
Scopusで見るGoogle Scholar
[47]
M. サクセナ、G.イェレッツィアン
NOD-Likeレセプター: 炎症と癌のマスターレギュレーター
フロント・イミュノール, 5 (2014)
グーグル スカラー
[48]
S.K. Kuss-Duerkop, A.M. Keestra-Gounder
多様な刺激によるNOD1およびNOD2の活性化:病原体が誘発する小胞体ストレスの感知に関わる役割の可能性
インフェクトイミュン, 88 (19) (2020), Article e00898
グーグルスカラー
[49]
J.D. Schertzer, A.K. Tamrakar, J.G. Magalhães, S. Pereira, P.J. Bilan, M.D. Fullerton, et al.
NOD1アクチベーターは自然免疫とインスリン抵抗性を結びつける
糖尿病, 60 (2011), pp.2206-2215
CrossRefView in ScopusGoogle Scholar
[50]
A.K. Tamrakar, J.D. Schertzer, T.T. Chiu, K.P. Foley, P.J. Bilan, D.J. Philpott, et al.
NOD2活性化により筋細胞自律的な自然免疫反応とインスリン抵抗性が誘導される
エンドクリノロジー, 151 (2010), pp.5624-5637
CrossRefView in ScopusGoogle Scholar
[51]
K.L. Chan, T.H. Tam, P. Boroumand, D. Prescott, S.R. Costford, N.K. Escalante, et al.
循環型NOD1活性化因子と造血型NOD1は代謝性炎症とインスリン抵抗性に寄与する
セル・レポート, 18 (2017), pp.2415-2426
PDFを見る記事を見るScopusGoogle Scholarで見る
[52]
S.L. リバース、A. クリップ、A. ジアッカ
NOD1: 自然免疫とインスリン抵抗性の間のインターフェイス
エンドクリノロジー, 160 (2019), pp.1021-1030
CrossRefScopusGoogle Scholarで表示する。
[53]
A. Sharma, S. Singh, A. Mishra, A.K. Rai, I. Ahmad, S. Ahmad, et al.
高脂肪食飼育マウスにおけるインスリン抵抗性とNOD1の漸増の対応関係
エンドクリン, 76 (2022), pp.282-293
CrossRefView in ScopusGoogle Scholar
[54]
A. Sharma, C.K. Maurya, D. Arha, A.K. Rai, S. Singh, S. Varshney, et al.
Nod1を介した脂肪分解は、脂肪細胞におけるPKCδ-IRAK軸を介してジアシルグリセロールの蓄積と連続的な炎症を促進する
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1865 (2019), pp.136-146
PDFを見る記事を見るScopusGoogle Scholarで見る
[55]
W. Chi, D. Dao, T.C. Lau, B.D. Henriksbo, J.F. Cavallari, K.P. Foley, et al.
細菌ペプチドグリカンはNOD1を介して脂肪細胞の脂肪分解を刺激する
PLoS ONE, 9 (2014), 記事e97675
CrossRefView in ScopusGoogleスカラー
[56]
A. シャルマ、S・シン、S・アーマド、F・ガルザー、J・D・シャーツァー、A・K・タムラカー
NOD1活性化は脂肪細胞においてNOX1/4を介して酸化ストレスを誘導する
フリーラジカルバイオロジー&メディシン, 162 (2021), pp.118-128
PDFを見る記事を見るScopusGoogle Scholarで見る
[57]
J.D. シャーツァー、A. クリップ
インスリン抵抗性にNODを与える
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌代謝学, 301 (2011), pp.585-586
Google Scholar
[58]
Q. 張、潘、曾、鄭、王、沈、他。
腸管リゾチームが微生物からNod1リガンドを解放し、膵臓β細胞におけるインスリン輸送を指示する
セル・レス, 29 (2019), pp.516-532
CrossRefView in ScopusGoogleスカラー
[59]
K.P. Foley, S. Zlitni, B.M. Duggan, N.G. Barra, F.F. Anhê, J.F. Cavallari, et al.
腸内細菌叢は肥満マウスのインスリンクリアランスを障害する
分子代謝学, 42 (2020), 記事101067
PDFを見る記事を見るScopusGoogle Scholarで見る
[60]
F.F.アンヘー、N.G.バーラ、J.D.シャーツァー
グルコースは腸内細菌叢と宿主生理の共生関係を変化させる
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌・代謝、318 (2020), pp.e111-e116
グーグル スカラー
[61]
H. ファング、F.F.アンヘー、J.D.シャーツァー
食事性糖質は代謝性疾患から守る免疫と微生物叢を低下させる
細胞メタボリズム, 34 (2022), pp.1422-1424
PDFを見る記事を見るScopusGoogle Scholarで見る
[62]
J.D. Schertzer、T.K.T. Lam
腸とマイクロバイオームによるインスリンの末梢および中枢制御
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌・代謝、320 (2021), pp.e234-e239
グーグル スカラー
[63]
J.F. Cavallari, M.D. Fullerton, B.M. Duggan, K.P. Foley, E. Denou, B.K. Smith, et al.
ムラミルジペプチドベースのポストバイオティクスはIRF4を介して肥満誘発性インスリン抵抗性を緩和する
セルメタボリズム, 25 (2017), pp.1063-1074
Scopusで見るGoogle Scholar
[64]
J.F. Cavallari、N.T. Pokrajac、S. Zlitni、K.P. Foley、 B.D. Henriksbo、 J.D. Schertzer
肝細胞のNOD2が肝-腸軸に関与し、マウスの脂肪肝疾患における脂肪症、線維症および腸内細菌異常症から保護される
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌・代謝、319 (2020), pp.e305-e314
グーグル スカラー
[65]
J.F. Cavallari, N.G. Barra, K.P. Foley, A. Lee, B.M. Duggan, B.D. Henriksbo, et al.
NOD2のポストバイオティクスは、マウスの血糖値と代謝性炎症を改善するために、非造血性RIPK2を必要とする
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌・代謝、318 (2020), pp.e579-e585
グーグル スカラー
[66]
B.M. Duggan, J.F. Cavallari, K.P. Foley, N.G. Barra, J.D. Schertzer
RIPK2が肥満雄マウスのチロシンキナーゼ阻害剤に対するインスリン応答を規定する
エンドクリノロジー, 161 (2020), p. bqaa086
グーグルスカラー
[67]
B.M. Duggan, K.P. Foley, B.D. Henriksbo, J.F.Cavallari, A.K. Tamrakar, J.D. Schertzer
Ripk2のチロシンキナーゼ阻害剤は、細菌細胞壁を介した脂肪分解、炎症、血糖値異常を減衰させる
Sci Rep, 7 (2017), p. 1578
ScopusGoogleスカラーで見る
[68]
B.M.ダガン、A.M.シン、D.Y.チャン、J.D.シャーザー
ポストバイオティクスは脂肪細胞のIRF4に関与し、肥満時の性差による血糖値の変化を促進する
生理学レポート, 10 (2022), 記事e15439
Scopusで見るGoogle Scholar
[69]
N.G.バラ、B.D.ヘンリクスボ、F.F.アンヘー、J.D.シャルツァー
NLRP3インフラマソームは脂肪組織の代謝を制御する
生化学雑誌, 477 (2020), pp.1089-1107
CrossRefView in ScopusGoogle Scholar
[70]
J. ルーケンス、V.D.ディキシット、T.-D. Kanneganti
肥満に関連する炎症性疾患と自己免疫におけるインフラマソームの活性化
Discov Med, 12 (2011), pp.65-74
ScopusGoogle Scholarで見る
[71]
R. Pahwa, A. Singh, B. Adams-Huet, S. Devaraj, I. Jialal
メタボリックシンドローム患者の皮下脂肪組織におけるインフラマソーム活性の亢進
糖尿病/メタボリズム研究・レビュー, 37 (2021), p. e3383
Scopusで見るGoogle Scholar
[72]
L. Antonioli, D. Moriconi, S. Masi, D. Bottazzo, C. Pellegrini, M. Fornai, et al.
減量と血糖コントロールがインフラマソームシグナルに与える影響の違い
肥満, 28 (2020), pp.609-615
CrossRefScopusGoogle Scholarで見る
[73]
C.J. タック、R. スティエンストラ、L.A.B. ヨーステン、M.G. ネテア
過剰脂肪とインスリン抵抗性を結びつける炎症:インターロイキン1ファミリーの役割
Immunol Rev, 249 (2012), pp.239-252
ScopusGoogleスカラーで見る
[74]
N. Esser、L. L'homme、A. De Roover、L. Kohnen、A. J. Scheen、M. Moutschen, et al.
肥満の表現型は、内臓脂肪組織のNLRP3インフラマソーム活性と免疫学的プロファイルに関連している
Diabetologia, 56 (2013), pp.2487-2497
CrossRefView in ScopusGoogle Scholar
[75]
A.J. Murphy, M.J. Kraakman, H.L. Kammoun, D. Dragoljevic, M.K.S. Lee, K.E. Lawlor, et al.
NLRP1インフラムソームからのIL-18産生は肥満とメタボリックシンドロームを予防する
セルメタボリズム, 23 (2016), pp.155-164
PDFを見る記事を見るScopusGoogle Scholarで見る
[76]
J. Salazar-León, A.L. Valdez-Hernández, S. García-Jiménez, L. Román-Domínguez, E. Huanosta-Murillo, L.C. Bonifaz, et al.
Nlrp1b1はIL-18産生を促進することで肥満誘発性炎症をネガティブに調節する
Sci Rep, 9 (2019), 記事 13815
Scopusで見るGoogle Scholar
[77]
M.J. McBride, K.P. Foley, D.M. D'Souza, Y.E. Li, T.C. Lau, T.J. Hawke, et al.
老齢マウスのサルコペニアと筋解糖能の低下にNLRP3インフラソームが寄与していること
アメリカン・ジャーナル・オブ・フィジオロジー-内分泌・代謝、313(2017)、pp.e222-e232
グーグルスカラー
[78]
M. フレスノ、R.アルバレス、N.クエスタ
Toll様受容体、炎症、代謝、肥満
生理学と生化学のアーカイブス, 117 (2011), pp.151-164
CrossRefView in ScopusGoogle Scholar
[79]
S. 明, 武田紘一
Toll様受容体のシグナル伝達
Nat Rev Immunol, 4 (2004), pp.499-511
CrossRefScopusGoogle Scholarで表示する。
[80]
V. 伊織、A.M.Iyer、T.Ravizza、L.Beltrame、L.Paracchini、S. Marchini、et al.
IL-1R1/TLR4経路の遮断は、後天性てんかんモデルにおける疾患修飾治療効果を媒介する
疾病の神経生物学, 99 (2017), pp.12-23
PDFを見る記事を見るScopusGoogle Scholarで見る
[81]
Y. Ji, S. Sun, N. Shrestha, L.B. Darragh, J. Shirakawa, Y. Xing, et al.
トール様受容体TLR2およびTLR4は、食事誘発性肥満における膵臓β細胞の複製を阻害する
Nat Immunol, 20 (2019), pp.677-686
CrossRefScopusGoogle Scholarで見る
[82]
A.C.ケーナー、J.C.ブリューニング
Toll様受容体:炎症と代謝をつなぐ
内分泌学・代謝学の動向, 22 (2011), pp.16-23
PDFを見る記事を見るScopusGoogle Scholarで見る
[83]
F.F. Anhê, N.G. Barra, J.F. Cavallari, B.D. Henriksbo, J.D. Schertzer
代謝性内毒素血症はリポ多糖の種類によって決定される
セル・レポート, 36 (2021), 記事 109691
PDFを見る記事を見るScopusGoogle Scholarで見る
[84]
H. リャン、S.E.ハッシー、A.サンチェス-アビラ、P.タンティウォン、N.ムジ
ヒト筋肉における炎症とインスリン作用に対するリポポリサッカライドの影響
PLoS ONE, 8 (2013), 記事e63983
CrossRefView in ScopusGoogle Scholar
[85]
L. Jia, C.R. Vianna, M. Fukuda, E.D. Berglund, C. Liu, C. Tao, et al.
肝細胞Toll様受容体4は、肥満誘発性炎症とインスリン抵抗性を制御する
Nat Commun, 5 (2014), p. 3878
Scopusで見るGoogle Scholar
[86]
J.H. Fritz, S.E. Girardin, C. Fitting, C. Werts, D. Mengin-Lecreulx, M. Caroff, et al.
Toll様受容体4とNOD1およびNOD2活性化アゴニストによるヒト単球および樹状細胞の相乗的刺激
Eur J Immunol, 35 (2005), pp.2459-2470
CrossRefView in ScopusGoogle Scholar
[87]
D.A. van Heel, S. Ghosh, M. Butler, K. Hunt, B.M.J. Foxwell, D. Mengin-Lecreulx, et al.
NOD1活性化によるToll様受容体反応の相乗的な増強
Eur J Immunol, 35 (2005), pp.2471-2476
CrossRefView in ScopusGoogle Scholar
[88]
L. ブーテンス、R.スティエンストラ
脂肪組織マクロファージ:肥満時の軌道修正
Diabetologia, 59 (2016), pp.879-894
CrossRefScopusGoogle Scholarで見る
[89]
K. Man, M. Miasari, W. Shi, A. Xin, D.C. Henstridge, S. Preston, et al.
転写因子IRF4は、TCR親和性を介した代謝プログラミングとT細胞のクローン拡大に必須である
Nat免疫学, 14 (2013), pp.1155-1165
CrossRefView in ScopusGoogle Scholar
[90]
Y. Lin、M. Bai、S. Wang、L. Chen、Z. Li、C. Li、et al.
乳酸は脂肪組織からのサイトカイン産生を調節することにより肥満とインスリン抵抗性を結びつける重要なメディエーターである
糖尿病, 71 (2022), pp.637-652
CrossRefView in ScopusGoogleスカラー
[91]
J. 江口、X. Wang、S. Yu、E.E. Kershaw、P.C. Chiu、J. Dushay, et al.
IRF4による脂肪の脂質ハンドリングの転写制御
細胞メタボリズム, 13 (2011), pp.249-259
PDFを見る記事を見るScopusGoogle Scholarで見る
[92]
J. Amar, R. Burcelin, J.B. Ruidavets, P.D. Cani, J. Fauvel, M.C. Alessi, et al.
見かけ上健康な男性におけるエネルギー摂取量と内毒素血症の関連性
アメリカ臨床栄養学会誌, 87 (2008), pp.1219-1223
PDFを見る記事を見るScopusGoogle Scholarで見る
[93]
A.B. Berezow, R.K. Ernst, S.R. Coats, P.H. Braham, L.M. Karimi-Naser, R.P. Darveau
Porphyromonas gingivalisとBacteroidesの構造的に類似したペンタアシル化リポ多糖は、顕著に異なる自然免疫応答を誘発する
微生物病態学, 47 (2009), pp.68-77
PDFを見る記事を見るScopusGoogle Scholarで見る
[94]
F.F. Anhê、A. Bhatwa、J.D. Schertzer
マウスにおけるポストバイオティクスの代謝的影響の決定
STAR プロトコル, 3 (2022), 記事 101098
PDFを見る記事を見るScopusGoogle Scholarで見る
[95]
F. Bäckhed, S. Normark, E.K.H. Schweda, S. Oscarson, A. Richter-Dahlfors
TLR4を介したLPSシグナル伝達の構造的要件:LPS修飾の生物学的役割
微生物と感染, 5 (2003), pp.1057-1063
PDFを見る記事を見るScopusGoogle Scholarで見る
[96]
F. ディ・ロレンツォ、A・シリポ、I・ビアンコーニ、N・I・ロレ、A・スカンポリーノ、L・スチュリアーレ、他。
持続性嚢胞性線維症分離株Pseudomonas aeruginosa RP73株は、低炎症活性の原因となるアシル化されていないLPS構造を示す
分子免疫学, 63 (2015), pp.166-175
PDFを見る記事を見るScopusGoogle Scholarで見る
[97]
B.M. Duggan, A.K. Tamrakar, N.G. Barra, F.F. Anhê, G. Paniccia, J.G. Wallace, et al.
腸内細菌叢に基づくワクチン接種は自然免疫に働きかけ、肥満マウスの血糖コントロールを改善する
分子代謝学, 55 (2022), 記事 101404
PDFを見る記事を見るScopusGoogle Scholarで見る
[98]
C. Pomié, V. Blasco-Baque, P. Klopp, S. Nicolas, A. Waget, P. Loubières, et al.
腸内常在菌による適応免疫系のトリガーがインスリン抵抗性と血糖異常から保護する
分子代謝学, 5 (2016), pp.392-403
PDFを見る記事を見るScopusGoogle Scholarで見る
[99]
M. ヴィジャイ・クマール、F.A.カルヴァーリョ、J.D.エイトケン、N.H.フィファダラ、A.T.ゲヴィルツ
フラジェリンによる体液性免疫の促進には、TLR5またはNLRC4が必要かつ十分な役割を果たす
Eur J Immunol, 40 (2010), pp.3528-3534
CrossRefView in ScopusGoogle Scholar
[100]
H.Q. Tran, R.E. Ley, A.T. Gewirtz, B. Chassaing
フラジェリン誘導型適応免疫は、フラジェリン微生物叢を抑制し、慢性炎症性疾患に対するワクチンとなる
Nat Commun, 10 (2019), p. 5650
Scopusで見るGoogle Scholar
引用者:(0)さん
Chang Gung Universityの責任で査読。
© 2023 Chang Gung University. 出版サービス:Elsevier B.V.
ScienceDirectについて
リモートアクセス
ショッピングカート
広告掲載
お問い合わせ・サポート
ご利用条件
個人情報保護方針
当社は、サービスの提供や強化、コンテンツや広告のカスタマイズのためにCookieを使用しています。継続することで、Cookieの使用に同意することになります。
Copyright © 2023 Elsevier B.V.またはそのライセンサーもしくは寄稿者。ScienceDirect® はElsevier B.V.の登録商標です。
この記事が気に入ったらサポートをしてみませんか?