
PyhtonのRDKitを使って,置換基をスクリーニングするDFTxTB計算用のinputを作る方法

PyhtonのRDKitのrdChemReactionsという模擬反応機能?を応用して,置換基をスクリーニングします。
反応の記述法は種々ブログやHPなどで紹介されているので割愛します。
例えば,ピロールの置換基をスクリーニングしたいときに,いきなり全通りやってもいいのですが,一旦臭素化して重複する可能性のある置換位置を消すやり方が,種々ブログやHPなどで紹介されています。
以下,順を追って説明すると,
AllChem.ReactionFromSmarts("[ch:1]>>[ch0:1]Br")でブロモ化反応を定義し、rxnと定義します。
Chem.MolFromSmiles("C1=CC=CN1")でtestにピロールをsmiles形式で入力してmol形式で定義します。
list(chain.from_iterable(rxn.RunReactants([test])))でtestに対してモノブロモ化を実施しリストに生成物を並べてmonobromidesとして定義します。
Draw.MolsToGridImageでmonobromides内の分子を描画します。
余談ですが,Draw.rdDepictor.SetPreferCoordGen(True)は,n員環のn ≧ 7の構造を描画する際に比較的綺麗に描画できる機能になります。
from rdkit import Chem
from rdkit.Chem import AllChem, Draw
from itertools import chain
rxn = AllChem.ReactionFromSmarts("[ch:1]>>[ch0:1]Br")
test = Chem.MolFromSmiles("C1=CC=CN1")
monobromides = list(chain.from_iterable(rxn.RunReactants([test])))
Draw.rdDepictor.SetPreferCoordGen(True)
Draw.MolsToGridImage(monobromides)
ちなみに,AllChem.ReactionFromSmarts("[ch:1]>>[ch0:1]Br")や,rdChemReactions.ReactionFromSmarts(rxnSmarts)で,rxnに定義した反応を描画できます。
また,反応は"[c:1]>>[c:1]Br"としても実用上問題はありません。
from rdkit.Chem import Draw
from rdkit.Chem import rdChemReactions
rxnSmarts = "[ch:1]>>[ch0:1]Br"
rxn = rdChemReactions.ReactionFromSmarts(rxnSmarts)
rxn
rxn2 = rdChemReactions.ReactionFromSmarts(rxnSmarts)
rxn2.Initialize()
rxn2rxnSmarts = "[c:1]>>[c:1]Br"
rxn3 = rdChemReactions.ReactionFromSmarts(rxnSmarts)
rxn3
生成した分子群はChem.MolToSmiles(mol) for mol in monobromidesでmonobromides内の分子をSMILESに変換した際に集合に入れることで重複を削除できます。ただ,分子が大きくなってきたときにあまり上手くワークしなかったので,より良い方法を模索中です。
再度molオブジェクトに変換した処理後のmolオブジェクトを格納したリストをmonobromides_uniqueと定義します。
上図ではピロールの向きもランダムなので,ついでに整列させるために,AllChem.Compute2DCoords(test)でtestの構造の二次元座標を計算します。
AllChem.GenerateDepictionMatching2DStructure(mol, test)でtestの構造をテンプレートとしてproducts_unique内のmolオブジェクトを整列します。
monobromides_unique内の分子を描画します。
monobromides_unique = [Chem.MolFromSmiles(smile) for smile in {Chem.MolToSmiles(mol) for mol in monobromides}]
AllChem.Compute2DCoords(test)
for mol in monobromides_unique:
if mol.HasSubstructMatch(test):
AllChem.GenerateDepictionMatching2DStructure(mol, test)
Draw.MolsToGridImage(monobromides_unique)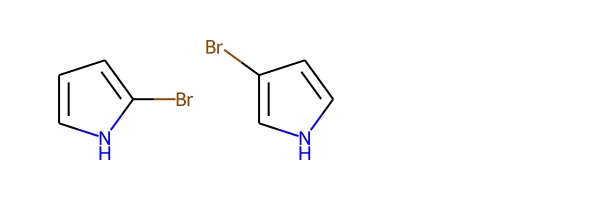
今回は上のピロールの臭素化体(ブロモピロール)に対して種々のヘテロアリール基を置換してみたいと思いますが、まずは、単純な例として上のブロモピロールの臭素置換基をピロールに置換します。
前述の変換反応をrxn_pyrroleと定義しました。
from rdkit.Chem.Draw import IPythonConsole
rxn_pyrrole = AllChem.ReactionFromSmarts("[Br:2][c:1]>>[c:1]C1=CC=CN1")
rxn_pyrrole.Initialize()ピロールに対して種々のヘテロアリール基を置換してみたいと思いますが、まずは、上のブロモピロールの臭素置換基をピロールに置換します。
最初に,Pys = []でピロール置換体を入れるための空のリストを作成しておきます。
今回はfor bromide_1st in monobromides_unique:で,上で作成したmonobromides_uniqueというリスト内の2つのピロールの臭素化体を1つずつ取り出して,Pys.extend(list(chain.from_iterable(rxn_pyrrole.RunReactants([bromide_1st]))))によって,rxn_pyrroleを行い,リスト化して,Pys.extendでPysの中に追加していきます。
Pys = []
for bromide_1st in monobromides_unique:
Pys.extend(list(chain.from_iterable(rxn_pyrrole.RunReactants([bromide_1st]))))
Pys = [Chem.MolFromSmiles(smile) for smile in {Chem.MolToSmiles(mol) for mol in Pys}]
#重複分子の削除
Draw.MolsToGridImage(Pys, molsPerRow = 5)
#monobromides内の分子を描画した。
続いて,ピロール上の窒素に対して種々のヘテロアリール基を一挙に置換してみたいと思いますが、考え方としては,さきほどとは逆に,まずは、ヘテロアリールを臭素化し,その臭素置換基をピロールに置換します。
今回はヘテロアリールは事前に準備してsdfとしてまとめてあるものを使います。
globで指定したディレクトリにあるsdfを参照してfilesに入れます。
import glob
import os
from os import chdir
import pandas as pd
chdir('/Users/NAME/Desktop/substituent_screening/HetAr')
files = glob.glob("./*.sdf")
for file in files:
print(file)
suppl = Chem.SDMolSupplier(file, removeHs=False)
mols = [mol for mol in suppl if mol is not None]
print(len(mols))./NX_HetAr.sdf
9
./S_HetAr.sdf
3
./O_HetAr.sdf
3
./N_HetAr.sdf
7
中身の例は以下の通り


まず,臭素化とピロールへの変換反応を定義します。
rxn_Br = AllChem.ReactionFromSmarts("[ch:1]>>[ch0:1]Br")
rxn_Br.Initialize()
rxn_Py = AllChem.ReactionFromSmarts("[Br:2][c:1]>>[c:1]N1C=CC=C1")
rxn_Py.Initialize()さらに反応後の分子をsdfとして保存したり,量子化学計算用のinputを作るために,mol_to_headerとmol_to_3dを定義します。
mol_to_headerでは,Chem.AddHsで水素を追加したり,elements = []以下で原子を数えています。
def mol_to_header(mol):
mol = Chem.AddHs(mol)
elements = []
for atom in mol.GetAtoms():
elements.append(atom.GetSymbol())
atom_num = len(elements)
header = str(atom_num) + "\n\n"
return headermol_to_3dでは,水素を追加した後,AllChem.EmbedMolecule(mol)で,molオブジェクトの3次元構造を生成して,AllChem.MMFFOptimizeMolecule(mol)でメルク分子力場を使って構造最適化しています。最近ではETKDGの方が速くて精度もいいという話もあるので,その場合は
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol)
を
p = AllChem.ETKDGv3()
AllChem.EmbedMolecule(mol, p)
で置き換えます。
その後のatoms = pd.DataFrame(columns=['element', 'x', 'y', 'z'])以降は,3次元構造をxyz形式で保存するための操作です。
def mol_to_3d(mol):
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol)
atoms = pd.DataFrame(columns=['element', 'x', 'y', 'z'])
elements = []
for atom in mol.GetAtoms():
elements.append(atom.GetSymbol())
for c in mol.GetConformers():
positions = c.GetPositions()
try:
atoms['element'] = elements
atoms['x'] = positions[:, 0]
atoms['y'] = positions[:, 1]
atoms['z'] = positions[:, 2]
except:
print("XYZError")
return atoms
先ほど参照したsdfのパスをまとめたfilesからsdfを順に参照しています。
num = 0は後で,作成したファイルを各フォルダに入れる際に番号で振り分けるためにファイルを参照する最初にnum = 0としています。
sdfからsuppl取り出した分子は,molsにまとめておきます。
これまでと同様に,Pys = []でピロール置換体を入れるための空のリストを作成しておき,molsから1分子ずつ取り出して,これまで通りの手順で臭素化とピロールへの変換を行い,Pys.extendで,Pysの中に追加していきます。
ちなみに,UpdatePropertyCacheというおまじないは,原子の原子価を計算して,許容値よりも高い価数の原子には例外を生成してくれます。
この処理はいつも行っておいた方が無難です。
writer = Chem.SDWriter('Py' + str(file.replace('.sdf','')) + ".sdf")で,作成した分子群をsdfで保存するファイル名を指定しています。
続いて,Pysから1分子ずつ取り出して,前述の通り,atoms = mol_to_3d(Py)で3次元構造を生成してxyz形式にして,header = mol_to_header(Py)で,xyz形式のファイルに必要なヘッダーを作っています。
writer.write(Py)で,上で指定したsdfファイルに書き出します。
ここからは、1分子ごとに番号を増やしてディレクトリを作成してその中に同名のpngとxyzファイルを保存していきます。
pngで保存する際は,平面(2D)の方が見やすいので,AllChem.Compute2DCoords(Py)で前処理してから,image = Draw.MolToFile(Py, str(file.replace('.sdf','')) + str(num) + '.png', size=(128, 128))で,pngとして分子の画像を保存しています。
最後に,with open(str(file.replace('.sdf','')) + str(num) + '.xyz', 'w') as gin:
で空のxyzファイルを作成して開き,gin.write(header)でヘッダーを書き出して, for v in atoms.values:以降で,atomsの構造情報を取り出して, gin.write(' '.join(map(str, v)) + '\n')で書き出しています。
おまじないのgin.write(' ' + '\n')
おまけで,with open('joblist.sh', mode = 'a') as gin_list:以降で,bashでジョブを一気に流せるようにjobのサブミットコマンドも書き出しています。
writer.close()で開いたものは必ず閉じる。
for file in files:
num = 0
chdir('/Users/NAME/HetAr/sdf/sustituents')
suppl = Chem.SDMolSupplier(file, removeHs=False)
mols = [mol for mol in suppl if mol is not None]
Pys = []
for mol in mols:
monobromides = list(chain.from_iterable(rxn_Br.RunReactants([mol])))
monobromides_unique = [Chem.MolFromSmiles(smile) for smile in {Chem.MolToSmiles(mol) for mol in monobromides}]
AllChem.Compute2DCoords(mol)
for monobromide in monobromides_unique:
if monobromide.HasSubstructMatch(mol):
AllChem.GenerateDepictionMatching2DStructure(monobromide, mol)
monobromide.UpdatePropertyCache(strict = False)
if rxn_Py.IsMoleculeReactant(monobromide):
Pys.extend(list(chain.from_iterable(rxn_Py.RunReactants([monobromide]))))
Pys = [Chem.MolFromSmiles(smile) for smile in {Chem.MolToSmiles(mol) for mol in Pys}]
file = file.replace('./','')
chdir('/Users/NAME/Desktop/substituent_screening/HetAr/Py')
writer = Chem.SDWriter('Py' + str(file.replace('.sdf','')) + ".sdf")
for Py in Pys:
atoms = mol_to_3d(Py)
header = mol_to_header(Py)
Chem.AddHs(Py)
AllChem.EmbedMolecule(Py)
AllChem.MMFFOptimizeMolecule(Py)
Py_NH = Chem.MolFromSmiles("[H]N1C=CC=C1")
AllChem.Compute2DCoords(Py_NH)
writer.write(Py)
num = num +1
os.makedirs('./' + str(file.replace('.sdf','')) + str(num), exist_ok=True)
chdir('/Users/NAME/Desktop/substituent_screening/HetAr/Py/' + str(file.replace('.sdf','')) + str(num))
if Py.HasSubstructMatch(Py_NH):
AllChem.Compute2DCoords(Py)
AllChem.GenerateDepictionMatching2DStructure(Py, Py_NH)
image = Draw.MolToFile(Py, str(file.replace('.sdf','')) + str(num) + '.png', size=(128, 128))
with open(str(file.replace('.sdf','')) + str(num) + '.xyz', 'w') as gin:
gin.write(header)
for v in atoms.values:
gin.write(' '.join(map(str, v)) + '\n')
gin.write(' ' + '\n')
chdir('/Users/NAME/Desktop/substituent_screening/HetAr/Py')
with open('joblist.sh', mode = 'a') as gin_list:
gin_list.write('cd ' + str(file.replace('.sdf','')) + str(num) + '\n')
gin_list.write('bsub -n 16 xtb ' + str(file.replace('.sdf','')) + str(num) + '.xyz --opt \n')
gin_list.write('cd .. \n')
writer.close()仕上がりはこんな感じです。
ここから先は
¥ 100
この記事が気に入ったらサポートをしてみませんか?