
ANCOM-BC2を用いた微生物群集構成の調査

微生物生態学では、微生物群集の環境変化による影響を理解することが重要です。
今回はQiime2で得られるファイルから、R上でANCOM-BC2という統計的手法を用いて、具体的に何の菌が変化したのかを調査します。
ANCOM-BC2(2023)はANCOM(2015)を改良した手法であり、バイアス補正とFDR制御のよりロバストな方法を持っているそう。詳しくは以下の元論文を参照してください。
私は最初にANCOMの方を使用したのですが、Qiime2に統合されており簡単に使用できる反面、変化した菌の検出が少なかったため、ANCOM-BC2も使用してみました。
実行環境とソフト
実行環境:Ubuntu 22.04.2 LTS (GNU/Linux 5.15.146.1-microsoft-standard-WSL2 x86_64)
実行ソフト:QIIME 2 2023.9 Amplicon Distribution上のR version 4.2.2
※WSL上でQiime2をactivateし、RでR環境を立ち上げています。
conda activate qiime2-amplicon-2023.9
Rデータの準備
はじめに、Qiime2での16S rRNAシーケンシングで得られた微生物群集データを読み込みます。
必要なライブラリをインストール・読み込みます。
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("phyloseq")
library(phyloseq)
if (!requireNamespace("devtools", quietly = TRUE)){install.packages("devtools")}
devtools::install_github("jbisanz/qiime2R")
library(qiime2R)taxonomy.qzaファイル、rooted-tree.qzaファイル、metadataファイル(tsv形式)、table.qzaファイルが必要です。
# taxonomyデータを読み込む
taxonomy <- read_qza("qzv_data/Taxonomy/taxonomy.qza")
taxtable <- taxonomy$data %>% as_tibble() %>% separate(Taxon, sep=";", c("Kingdom","Phylum","Class","Order","Family","Genus","Species"))
# 系統樹データを読み込む
tree <- read_qza("rooted-tree.qza")
# サンプルメタデータを読み込む
metadata <- read.table("sample-metadata.tsv", sep='\t', header=T, comment="")
# OTUテーブルを読み込む
otu_unrarefied <- read_qza("qzv_data/Table/table.qza")
# phyloseqオブジェクトの作成
physeq_unrarefied <- phyloseq(
otu_table(otu_unrarefied$data, taxa_are_rows = T),
phy_tree(tree$data),
tax_table(as.data.frame(taxtable) %>% select(-Confidence) %>% column_to_rownames("Feature.ID") %>% as.matrix()),
sample_data(metadata %>% as.data.frame() %>% column_to_rownames("sample.id"))
)メタデータファイル
> head(metadata)
SampleID 3groups 4groups days-since-experiment-start date-taken
1 imm2rpt imm imm 0 231116
2 imm3rpt imm imm 0 231116
3 imm4rpt imm imm 0 231116
4 1day3 1day 1day 1 231030
5 1day4 1day 1day 1 231030
6 1dayrpt 1day 1day 1 231116サンプルサイズの違いによるを考慮し公平な比較とするため、"Feature Count"(table.qzvをQiime2 Viewで見ると分かる)の値の最小値でデータを精製します。
最小値が他のサンプルとあまりにかけ離れている場合などは適宜適切な値を選択してください。
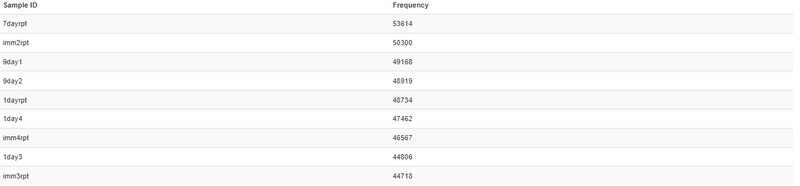
# データの精製
physeq_rarefy <- rarefy_even_depth(physeq_unrarefied, sample.size = 44718)ANCOM-BC2解析
データが準備できたら、ANCOM-BC2解析に進みます。TreeSummarizedExperimentオブジェクトを作成し、ANCOMBCライブラリをインストールします。
# PhyloseqオブジェクトをTreeSummarizedExperimentオブジェクトに変換
tse <- mia::makeTreeSummarizedExperimentFromPhyloseq(physeq_rarefy)
# ANCOMBCライブラリをインストールしロード
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ANCOMBC")
library(ANCOMBC)"Update all/some/none?[a/s/n]と表示された場合は、nで基本的にokです。
グループの順番を決めます。2グループで変化した菌を決定する操作になるため、最初の1グループとの比較になります。今回の場合はimmグループと1dayグループ、immグループとlongグループの結果が出力されます。
# グループの順番を決める
tse$X3groups <- factor(tse$X3groups, levels = c("imm", "1day", "long"))ANCOM-BC2解析を実行します。変数がたくさんありますが、詳しくは以下リンクの説明参照してください。筆者にも理解できない変数が多かったです。。
https://github.com/FrederickHuangLin/ANCOMBC/blob/bugfix/man/ancombc2.Rd
重要なものだけ紹介します。
"tax_level"は生物学の階級を指定しており、界"Kingdom"、門"Phylum"、綱"Class"、目"Order"、科"Family"、属"Genus"、種"Species"の7つから選択できます。種の方に近づくほど、データ量が多くなり時間がかかります。
"fix_formula"と"group"には比較対象の列名を入れてください。
set.seed(123)
# ANCOM-BC2解析
output <- ancombc2(data = tse, assay_name = "counts", tax_level = "Family",
fix_formula = "X3groups",
rand_formula = NULL,
p_adj_method = "holm", pseudo_sens = TRUE,
prv_cut = 0.10, lib_cut = 1000, s0_perc = 0.05,
group = "X3groups", struc_zero = TRUE, neg_lb = TRUE,
alpha = 0.05, n_cl = 2, verbose = TRUE,
global = TRUE, pairwise = TRUE, dunnet = TRUE, trend = TRUE,
iter_control = list(tol = 1e-2, max_iter = 20,
verbose = TRUE),
em_control = list(tol = 1e-5, max_iter = 100),
lme_control = lme4::lmerControl(),
mdfdr_control = list(fwer_ctrl_method = "holm", B = 100),
trend_control = list(contrast = list(matrix(c(1, 0, -1, 1),
nrow = 2,
byrow = TRUE)),
matrix(c(-1, 0, 1, -1),
nrow = 2,
byrow = TRUE),
matrix(c(1, 0, 1, -1),
nrow = 2,
byrow = TRUE),
node = list(2),
solver = "ECOS",
B = 100))結果をcsv形式で出力します。
# Write results to CSV files
write.csv(output$res, "ancombc_species/ancombc_prim_family.csv", row.names=FALSE)
write.csv(output$res_global, "ancombc_species/ancombc_global_species.csv", row.names=FALSE)
write.csv(output$res_pair, "ancombc_species/ancombc_pair_species.csv", row.names=FALSE)
write.csv(output$res_dunn, "ancombc_species/ancombc_dunn_species.csv", row.names=FALSE)
write.csv(output$res_trend, "ancombc_species/ancombc_trend_species.csv", row.names=FALSE)
bias_correct_log_table <- output$bias_correct_log_table
bias_correct_log_table[is.na(bias_correct_log_table)] <- 0
write.csv(round(bias_correct_log_table, 2), "ancombc_species/ancombc_biascorrectlogtable_species.csv", row.names=FALSE)結果考察
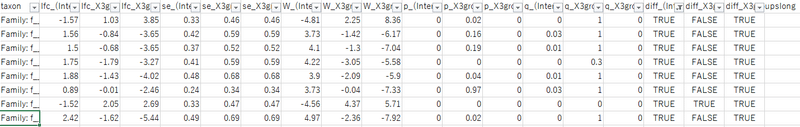
csvファイルをエクセルなどでフィルタして、diffがTRUEとなっているものが有意に差を持つ科です。lfcはlog fold changeの略で、正であれば基準のグループと比べ増加したことを表しています。
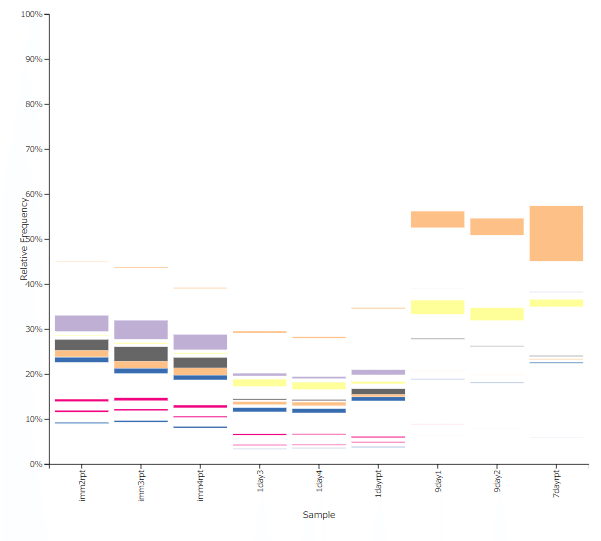
検出された科を、菌叢構成の棒グラフで見ると分かりやすいです。オレンジの科は増え、紫は減っている科です。
まとめ
有意な菌叢変化を検出する統計的な手法になります。
次回は機能解析の可視化についてです。
さらにANCOM-BC2の結果からボルケーノプロットを作成する記事も書きました。以下リンクより作ってみてください。
この記事が気に入ったらサポートをしてみませんか?