Qiime2とRを用いて菌叢の機能予測をする

微生物群集の機能分析は代謝ポテンシャルや菌叢の機能的多様性の知見を得ることに繋がります。
今回はQiime2で得られたデータとRを用いて菌叢解析を試みます。
実行環境とソフト
実行環境:Ubuntu 22.04.2 LTS (GNU/Linux 5.15.146.1-microsoft-standard-WSL2 x86_64)
実行ソフト:QIIME 2 2023.9 Amplicon Distribution上のR version 4.2.2
※WSL上でQiime2をactivateし、RでR環境を立ち上げています。
conda activate qiime2-amplicon-2023.9
Rライブラリとデータの準備
必要なパッケージをインストールし、ライブラリを読み込みます。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
pkgs <- c("phyloseq", "ALDEx2", "SummarizedExperiment", "Biobase", "devtools",
"ComplexHeatmap", "BiocGenerics", "BiocManager", "metagenomeSeq",
"Maaslin2", "edgeR", "lefser", "limma", "KEGGREST", "DESeq2")
for (pkg in pkgs) {
if (!requireNamespace(pkg, quietly = TRUE))
BiocManager::install(pkg)
}
library(readr)
library(ggpicrust2)
library(tibble)
library(tidyverse)
library(ggprism)
library(patchwork)メタデータファイルを読み込みます。Qiime2で一般的に用いられるメタデータファイルと形式が違う(2行目を削除)ので、コピーして修正してください。
metadata <- read_delim("sample-metadata_4r.txt", delim = "\t", escape_double = FALSE, trim_ws = TRUE) > head(metadata)
# A tibble: 6 × 3
`sample-id` `3groups` `4groups`
<chr> <chr> <chr>
1 imm2rpt imm imm
2 imm3rpt imm imm
3 imm4rpt imm imm
4 1day3 1day 1day
5 1day4 1day 1day
6 1dayrpt 1day 1day機能ポテンシャルの情報を含む、KEGGパスウェイデータを読み込みます。"qiime2 picrust2"で得られるファイルです。
kegg_abundance <- ko2kegg_abundance("q2-picrust2_output/ko_metagenome_exported/ko-feature-table.biom_copy.tsv") データの解析
グループ間での差異を示す経路を特定するため、統計解析であるDAA解析をします。daa_methodはALDEx2、DESeq2、Maaslin2、LinDA、edgeR、limma voom、metagenomeSeq、Lefserの中から選択します。groupにはmetadataで比較するグループの記入された列名を、referenceには基準とするグループ名を入力します。
daa_results_df <- pathway_daa(abundance = kegg_abundance, metadata = metadata, group = "3groups", daa_method = "LinDA", select = NULL, reference = "imm") KOからKEGGへの変換を使用してパスウェイの結果にアノテーションを付け、パスウェイの説明を追加します。
#LinDA解析したもののみをフィルタ
daa_sub_method_results_df <- daa_results_df[daa_results_df$method == "LinDA", ]
daa_annotated_sub_method_results_df <- pathway_annotation(pathway = "KO", daa_results_df = daa_sub_method_results_df, ko_to_kegg = TRUE)> head(daa_annotated_sub_method_results_df)
feature method group1 group2 p_values adj_method p_adjust
118 ko00121 LinDA 1day imm 0.000215855428786499 BH 0.014354386
154 ko04020 LinDA 1day imm 3.38378742678068e-06 BH 0.001800175
217 ko04512 LinDA 1day imm 0.000129242043539559 BH 0.009822395
353 ko00940 LinDA long imm 0.00010523835584281 BH 0.009789557
420 ko04020 LinDA long imm 1.47758305470152e-05 BH 0.002620247
432 ko05414 LinDA long imm 1.44884556164919e-05 BH 0.002620247データフレームを確認すると、group1が比較対象のグループなので、比較したいグループのみを抽出します。
daa_longfiltered_df<- daa_annotated_sub_method_results_df[daa_annotated_sub_method_results_df$group1 == "long", ]エラーバープロットの作成
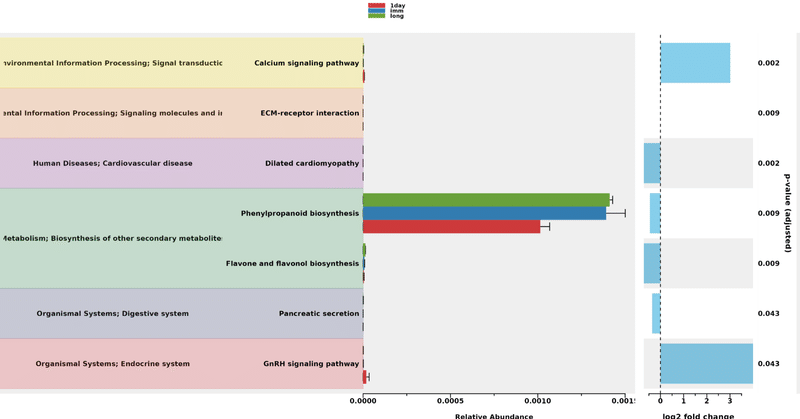
経路のエラーバープロットを生成し、グループ間の差異を可視化します。Groupをmetadata$比較するグループの列名としてください。
p <- pathway_errorbar(abundance = kegg_abundance, daa_results_df = daa_longfiltered_df, Group = metadata$'3groups', p_values_threshold = 0.05, order = "pathway_class", select = NULL, ko_to_kegg = TRUE, p_value_bar = TRUE, colors = NULL, x_lab = "pathway_name")
#pngの画像ファイルとして保存
ggsave("picrust2_linda_3groupslongfilter.png", height=9, width=16, device="png")
有意な差異を示した経路が視覚的に判明します。
ヒートマップの作成
全てのグループを比較するヒートマップになります。groupは比較したいグループの記述された列を指定してください。
#ggh4xパッケージをインストール
install.packages("ggh4x")
library("ggh4x")
#有意差を示す経路のみを抽出
feature_with_p_0.05 <- daa_annotated_sub_method_results_df %>% filter(p_adjust < 0.05)
#色の指定
custom_colors <- c("skyblue", "salmon")
#pathwayという名前の列を作る
kegg_abundance <- kegg_abundance %>%
tibble::rownames_to_column(var = "pathway")
#ヒートマップ pathway_heatmap(
abundance = kegg_abundance %>%
right_join(
daa_annotated_sub_method_results_df %>% select(all_of(c("feature","pathway_name"))),
by = c("pathway" = "feature")
) %>%
distinct(pathway, .keep_all = TRUE) %>%
filter(pathway %in% feature_with_p_0.05$feature) %>%
select(-"pathway") %>%
column_to_rownames("pathway_name"),
metadata = metadata,
group = "3groups",
colors = custom_colors
)
#pngの画像ファイルとして保存
ggsave("picrust2_linda_3groups_heatmap.png", height=9, width=16, device="png")

有意に差異を示した経路と、経路の関連遺伝子の多い・少ないグループが視覚的に表現されています。
まとめ
視覚的な図として使いやすいです。
ここではサンプル数が少なくなってしまいましたが、サンプル数が多いほどより信頼性の高いものとなります。
次回はANCOM-BC2の結果からボルケーノプロットを作成します。
この記事が気に入ったらサポートをしてみませんか?