RでWGCNA解析~解釈のためのヒートマップ作製とデータの出力~

前回まででWGCNAの基本的な操作は終えました。(ブロックごとのクラスタリングなど別の方法はありますが、それは本家本元のチュートリアルを見てもらえれば理解はできると思います。)
今回は各モジュール同士、及びモジュールと組織のヒートマップを作製し、各モジュールに入っている遺伝子のリストをCSVファイルとして出力するところまでやります。
ヒートマップの作製
例によって今まで書いたコードと地続きです。この記事の一番下に今までのコードをまとめて載せているので、コピペしてから始めてください。
#モジュールの第一主成分を抜き出し、ヒートマップを作れるようにする
MEList = moduleEigengenes(Data2, colors = dynamicColors)
MEs = MEList$eigengenes
#モジュール同士のヒートマップを作る。
#おすすめのヒートマップ作製ライブラリーなのでインストール推奨
library(corrplot)
#相関係数を計算する
corr <- cor(MEs, method = "pearson")
#表示
corrplot(corr, tl.col="black",type = "upper", method="color")
#モジュールと組織のヒートマップを作る
corr2<-t(as.matrix(MEs))
corrplot(corr2, tl.col="black", method="color")
以下出力結果。このヒートマップ関数の使いかたは別途調べてください。そんなに難しくないのと、割と見た目もきれいなのでおすすめです。

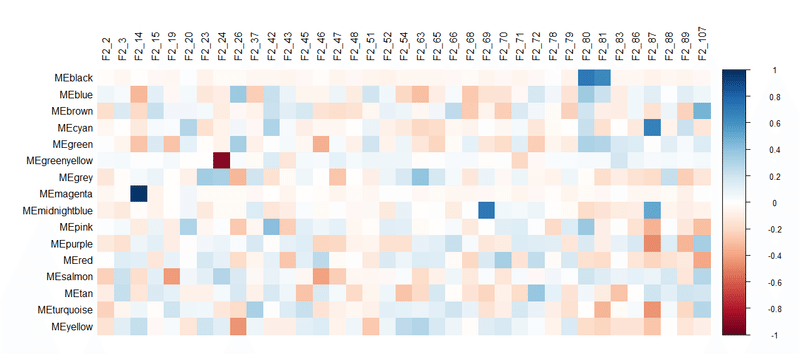
こうすることで特異的なモジュールを見つけやすくなります。
データの出力
各モジュールにどの遺伝子が割り当てられていたかを出力します。ここでは元データの一番右列にモジュールの情報を加える形を目指します。
#モジュールごとに遺伝子をまとめたCSVファイルを作成する
#モジュールの名前を遺伝子名に対応させる
newnames<-colnames(Data2)
names(dynamicColors)=newnames
#元データに結合して保存
Data = read.csv("LiverFemale3600.csv",header=T,row.names=1)
CompData<-cbind(Data,dynamicColors)
write.csv(CompData,"Modules.csv")これでモジュールの情報を元データに紐づけることができました。これを用いてパスウェイ解析などに移行することも、またメタボロームデータと統合解析することも可能です。
過去のコード
library(WGCNA);
options(stringsAsFactors = FALSE);
#データを行列として読み込み
Data1 = read.csv("LiverFemale3600.csv",header=T,row.names=1)
Data2<- t(as.matrix(Data1))
#ここの転置しないと組織ごとにモジュールを検出してしまいます
library(matrixStats)
library("dplyr")
library(igraph) #グラフの計算
library(visNetwork) #グラフの可視化
library(colorBlindness) #カラーパレットの取得
library(tidyverse)
#R^2値の計算
powers = c(1:30)
sft <- pickSoftThreshold(Data2, powerVector = powers, verbose = 5)
#計算した値のプロット
cex1 = 0.9;
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
abline(h=0.90,col="red")
#スケールフリー性を持つかどうかを見る
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
#まずは相関係数を重み付きで計算。前回の記事で10に設定したのでそれで行う。
softPower = 10;
adjacency = adjacency(Data2, power = softPower);
#データによってはここの計算に結構時間がかかる。気長に待ちましょう。
TOM = TOMsimilarity(adjacency);
dissTOM = 1-TOM
#樹形図の作成
geneTree = hclust(as.dist(dissTOM), method = "average");
#描写
sizeGrWindow(12,9)
plot(geneTree, xlab="", sub="", main = "Gene clustering on TOM-based dissimilarity",
labels = FALSE, hang = 0.04);
# モジュールサイズの最小値を決定します。一つのグループの最小の大きさで、小さいほどモジュールが細かく分かれる。
minModuleSize = 30;
# モジュールの特定
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM,
deepSplit = 2, pamRespectsDendro = FALSE,
minClusterSize = minModuleSize);
table(dynamicMods)
#モジュール番号を色に変換
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)
#表示
sizeGrWindow(8,6)
plotDendroAndColors(geneTree, dynamicColors, "Dynamic Tree Cut",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05,
main = "Gene dendrogram and module colors")この記事が気に入ったらサポートをしてみませんか?