
麻酔科医が知っておくべき心筋細胞の生理学

今回は、心筋細胞の生理学の入門についてです。
秋田大学の山本先生が書かれているhttps://jscvs.or.jp/surgery/0_3_syujutu_kaisinjutu/の記事をベースに改変したものとなっております。
JaSECTで講演されている内容でわからん!となっている方も、文字起こしされた本記事を見て理解を深めていただけるのではないでしょうか?
興奮収縮連関
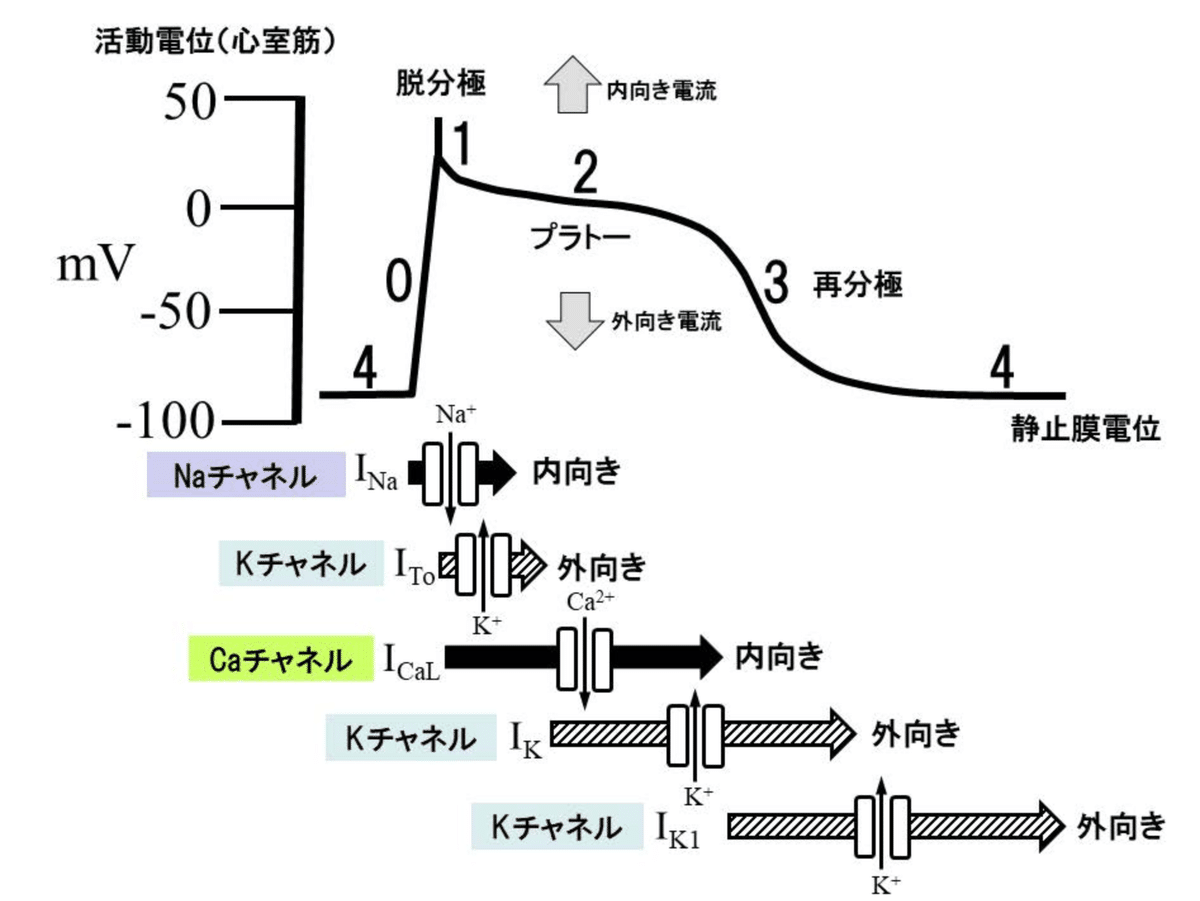
活動電位について
活動電位の発生から筋収縮/弛緩までの一連の電気生理学的反応である。
第0相では、電位依存性Na+チャンネル開口によって細胞内に急速にNa+が流入し(Na+電流、INa)脱分極が生じる。
第1相では、Na流入後に一過性のK+流出(ITo)による再分極によるスパイクが生じる。
第2相では、電位依存性L型Ca2+チャネルを介する長いCa2+流入(ILCa)によるプラトーが形成される。
第3相では、K+流出(遅延整流K+電流、IK)による再分極が生じて静止膜電位に向かう。
第4相では、高いK+透過性(内向き整流K+電流、IK1)と低いNa+透過性によって分極(細胞膜内側に負の電荷が残る)が維持されて静止膜電位が形成されている。
静止膜電位はその部位によって異なり、心室筋で-90mV、心房筋で-80mV ~-90mV。洞房結節や房室結節における静止膜電位はより浅く、洞房結節:-50mV ~-60mV、房室結節:-60mV~-70mVとなっている。
細胞内カルシウムハンドリングについて
興奮収縮連関における一連の細胞内Ca2+動態はカルシウムハンドリングと呼ばれている。心室筋細胞では活動電位(第0相)が生じると、電位変化はT管の電位依存性L型Ca2+チャネルを活性化させ細胞質へCa2+が流入する。細胞質へ流入したCa2+は、T管細胞膜に面している筋小胞体終末槽のCa2+チャネルを開口させ、筋小胞体内のCa2+が細胞質に放出される(CICR、Ca2+-induced Ca2+ releaseと呼ばれる)。細胞質Ca2+濃度が上昇するとATPを加水分解しながらアクチンとミオシンの相互作用(クロスブリッジおよびスライディング)を通じて筋収縮が生じる。筋小胞体のCa2+放出は一過性であり、その後、ATP依存性の筋小胞体Ca2+ポンプによるCa2+取り込みが優位となり細胞質Ca2+が低下し筋弛緩が生じる。活動電位とともに生じる細胞質Ca2+の一過性増減をCa2+トランジェントと呼ぶ。ミトコンドリアもまたCa2+貯蔵が可能であり、上昇した細胞質Ca2+は内膜にあるCa2+運搬体(Ca2+ uniporter)を介してマトリックスにも取り込まれ、クエン酸回路のCa2+依存性脱水素酵素の活性化に関与している。
心不全では心室の拡張不全と収縮不全が生じるが、拡張不全の原因は細胞内Ca2+ハンドリングの異常(筋小胞体リアノジン受容体の過リン酸化によるCa2+漏出と筋小胞体Ca2+ポンプの発現減少)により細胞質Ca2+除去能が低下し心筋弛緩の遅延が生じるとともに心肥大や線維化による心筋の硬さの増大による。また収縮不全の原因は、筋小胞体Ca2+貯留の低下による収縮時Ca2+動員の低下、筋原線維のCa2+依存性発生張力の低下で生じるが、アポトーシスによる細胞死や線維化もそれに関与する。心不全ではNa+/Ca2+交換機構の発現が増大しその逆モード(細胞外からのCa2+流入)が増強している。これは細胞内Ca2+ハンドリングの異常と相まって細胞質Ca2+量を恒常的に高い状態に維持している。圧負荷肥大心では心筋重量増大に比較して血管増殖能は低く、経時的に血管密度が低下する。


心筋細胞の代謝
通常の心筋のエネルギー基質は、主として60%が脂肪酸(β酸化)、30%がブドウ糖(解糖系→38molのATP産生)である。未熟心筋では解糖系に大きく依存しブドウ糖や乳酸をより利用している。ミトコンドリアではβ酸化や解糖系で産生されたアセチルCoAがクエン酸回路に入り、基質分子の水素が脱水素酵素により抜きとられる。そのうち電子は電子運搬体(NADHやFADH2)を介して内膜の電子伝達系に渡され、複合体間の酸化還元過程でプロトン(H+)が膜間腔に汲み上げられマトリックスとのH+濃度勾配が形成される(その際、約-180 mVの内膜電位が形成される)。そのH+濃度勾配を利用して共役するATP合成酵素(F1Fo-ATPase)がATPを合成する。これらの一連の過程は酸化的リン酸化と呼ばれる。ATPの高エネルギーリン酸(~P)は、クレアチンリン酸シャトルを介してミトコンドリアからクレアチンリン酸として細胞質に運ばれ、再びATPに変換され細胞機能を維持するため利用される。電子は最終的に酸素(電子受容体)に渡され水分子となる。クエン酸回路の酸化過程(酸化的脱炭酸)で生じたCO2は拡散によって細胞外へ排出される。虚血では酸素(電子受容体)が不足し酸化的リン酸化とクエン酸回路は停止する。虚血早期では解糖系が優勢になるが、細胞内の酸性化が進むと解糖系も停止する。
虚血再灌流障害
これには可逆性の心筋スタンニングと不可逆性の心筋細胞死の2つがあり、問題のない手術はだいたいが前者であり、心筋梗塞は後者である。
虚血が再灌流されると早期に過剰収縮が生じてから徐々に心機能が回復するが、心筋傷害の程度によって低い収縮期圧(収縮機能障害)と高い拡張期圧(拡張機能障害)が残存する。

虚血による心停止が一定時間を過ぎると、心筋の拘縮が生じる。虚血中の心筋拘縮は、ATPの低下によってミオシン線維へのATP結合が低下し、アクチンとミオシンのクロスブリッジが解除されない状態であることを示している。再灌流早期に見られる過剰な心筋収縮は再灌流の多彩な細胞内機序を反映し、低心機能残存の原因となる。その機序は"細胞内Ca2+過負荷"、"ATP低下"、"細胞内pHの急激な変化"、"活性酸素の発生"、によって特徴付けられる。
細胞内の酸性化とCa2+過負荷
ATP利用と解糖はH+を生成するため、虚血では細胞内の酸性化が進む。虚血によって細胞内H+の増加が緩衝系の能力を超えると、Na+/H+交換機構によるH+排出とNa+流入が生じ細胞内Na+濃度が上昇する。虚血中、細胞内H+によって抑制されているNa+/Ca2+交換機構が、再灌流時のH+洗い出し抑制が解除されるとNa+排出/Ca2+流入が生じ細胞内Ca2+過負荷に至る(図8)。細胞内ATPの推移はNa+/K+ポンプ活性に影響し、ATP濃度が低ければNa+/K+ポンプは細胞内Na+濃度低下への寄与が乏しい。また他の要因として細胞膜Ca2+チャネルを介するCa2+流入、筋小胞体Ca2+ポンプの抑制、補体活性亢進やラジカル発生等による傷害細胞膜を介するCa2流入も細胞内Ca2+過負荷の要因である。ミトコンドリアはCa2+の緩衝機能を有しており細胞質Ca2+過負荷が生じると、ミトコンドリア内膜のCa2+運搬体がCa2+を取り込み細胞質Ca2+を低下させようとする。細胞内Ca2+過負荷はミトコンドリアCa2+過負荷を来す。
活性酸素
活性酸素(スーパーオキシドラジカルやヒドロキシラジカル、過酸化水素)は酸素分子に対する不十分な電子供給過程で生じる。虚血再灌流による活性酸素の発生源としては、ミトコンドリア電子伝達系とヌクレオチド分解が重要である。ミトコンドリアでは、電子受容体である酸素が虚血時に欠乏し電子運搬体(NADHやFADH2)からの電子が電子伝達系内に停滞すること、および再灌流時におけるO2濃度の急激な上昇によって活性酸素が発生する。また虚血中のアデニンヌクレオチドの分解(ATP、ADP、AMP)から脱リン酸化と脱アミノ化を経てイノシンが生じた後、ヒポキサンチン→キサンチン→尿酸の過程で活性酸素種が発生する。ミトコンドリアにおいて発生した活性酸素種は電子伝達系からチトクロームCを漏出させ、カスパーゼ9を介してアポトーシスに致る。
過酸化水素は一電子還元を受けて酸化力が強いヒドロキシラジカルに変化し、膜リン脂質の酸化を経て脂質ラジカルが発生し、膜の自動過酸化による膜傷害に至る。
この記事が気に入ったらサポートをしてみませんか?