
座談会:次世代の生命情報科学に向けて 前編


荒川 和晴
慶應義塾大学 先端生命科学研究所
岩崎 渉
東京大学 大学院新領域創成科学研究科
大上 雅史
東京工業大学 情報理工学院 情報工学系
尾崎 遼
筑波大学 医学医療系/人工知能科学センター
小林 徹也
東京大学 生産技術研究所
福永 津嵩
早稲田大学 高等研究所
編集:尾崎・福永
今から10年前の2012年に、バイオインフォマティクスのあり方について、若手研究者による多様な議論が交わされたことがありました。
JSBiニュースレター第25号に「次世代の生命情報科学に向けて - 若手研究者からの視点・論点」と題する特集が組まれ、また、同年10月の生命医薬情報学連合大会において、「NGS 現場の会・オープンバイオ研究会・生命情報科学若手の会・定量生物学の会 4会合同シンポジウム『これからの生命科学を考える』」が開催されました。
それから10年経ち、この時の「答え合わせ」と今後の展望を議論するため、2022年1月31日に、2012年当時の著者・演者である荒川・岩崎・大上・小林と尾崎・福永で座談会をZoomで開催しました。
本特集では、その座談会の様子を前後編に分けてお届けいたします。後編は次号に掲載予定です。
配列相同性検索と遺伝子機能推定
尾崎:本日はお集まりいただきありがとうございます。今日の座談会の目的ですが、まず2012年に何があったかを簡単に説明させていただきます。
2012年は、若手研究者によるバイオインフォマティクスのあり方について、多様な議論が交わされていました。まずJSBiのニュースレターで『次世代の生命情報科学に向けて・若手研究者からの視点・論点』という特集があり、荒川さん、岩崎さん、大上さん、川島武士さん、佐藤行人さんが熱い記事を書かれていました。
また、その年の生命医薬情報学連合大会では、「これからの生命科学を考える」という題で、NGS現場の会・オープンバイオ研究会・生命情報科学若手の会・定量生物学の会の合同シンポジウムがありました。このシンポジウムではセッションが3つもあって、4会から12人の演者が参加し、様々な観点から今後の生命科学の方向性について議論が交わされていました。
今年2022年はそれからちょうど10年目なので、その「答え合わせ」をしたいと考えています。テーマとしては3つあります。
(1) 10年前に考えていて予想どおりだったこと
(2)10年前に考えていて結構変わったこと
(3)10年後どうなるか
を議論できればと思います。まずは過去から今までの話をできればと思います。
まず、岩崎さんから、10年前に考えていて予想どおりだったことと、結構予想と変わったことを話してもらってもいいでしょうか。
岩崎:私は、配列相同性検索(配列類似性検索)がボトルネックになってるのがしんどいなということは、この10年間ずっと思っていました(図1〜2)。これについては、AlphaFoldのように配列から構造を予測できる、配列だけから解析できることも進みつつあるので、そういう意味では、少し変わりつつあるのかなというふうに思ってはいます。


「ゲノム解読」という言葉は、昔はよく使われていましたけど、最近あまり使われてないような気がしていました。というのは、解読というからには、何か意味を読み取れないといけないと思うんですが、文字列を明らかにしても、そこから意味を読み解くということは、なかなか難しかったということがあったと思います。ただ最近だと、配列からいろんなことが、構造も予測できるし、どこの遺伝子がどれぐらい転写してるかとか、エピジェネティックな状態とかも予測できるようになってきたということで、ようやく、20年前からゲノム解読という言葉を使っていましたけど、そういうことが可能になってきたかなというふうにも思ってはいます。
尾崎:配列相同性検索でアノテーションできるものって増えたんですか。類似性検索をする場合に、本当に全然データベースに類似配列がないねっていう場合と、類似する配列はあっても、結局アノテーションが付いてない配列っていうのがあって知識が増えないという場合がありますよね。それはずっと昔からあると思うんですけど、アノテーションされてない配列の知識を精力的に増やす研究って、例えば、バクテリアだったり、モデル生物や人でもいいんですけど、あったりしましたっけ。
大上:私の個人的な意見だと、そういう一つ一つのアノテーションを、いわば、博物学的にやるっていう研究は、もちろん今までもこれからも続くと思うんですけど、「よく分かんないけど全体から何とか情報を抽出してやろう」みたいな研究は増えましたね。メタゲノムもまさにそうだと思いますけど、よく分かんないまま全体を扱って、その中からなんか意味ありげな雰囲気を醸し出している情報を抽出してやろうみたいな研究は増えて、たくさん多分出てきたんですけど、よく分からんっていうのが現状のところかなというふうに思ってます。
岩崎:メタボロームをいろんな条件で網羅的に測って、遺伝子の機能を予測しようみたいな研究は、荒川さんがつぶやいてませんでした?「これをやるべきだった」みたいな。
荒川:メタボロームでというのは、ちょっと分からないですけど、よく使われるのは、発現相関ですよね。どういうタイミングで同じように遺伝子発現が変化しているかということを調べて、大体類似しているパスウェイを探そうというようなスクリーニングは前からやられていて、それは配列の類似性は全く関係なくやっている。特に細胞の種類が多い多細胞生物だと、発現しているタイミングと、どういう細胞でどう局在するかというところが一致していれば、比較的似ているカテゴリーのものが多くなってくるだろうと類推するスクリーニングは、比較的有効に使われている技術の中の一つかなというふうには思います。
福永:結局CRISPRで各遺伝子をつぶしてみたいな研究は網羅的にやられてるわけで、そういう意味では、機能がよく分かってない遺伝子についても、何らかしらの実験結果は、探せば出てくるとは思うんですけど、それをどうまとめる、結局どの段階で既に機能が分かったっていうのがよく分からないというところはあるんじゃないかとは思いますけれども。
岩崎:バクテリアだとKeioコレクションは前からありますよね。
荒川:例えば、酵母とかで、ノックアウトしてもフェノタイプが変化しない遺伝子が半分ぐらいじゃないですか。こういうことは、もう20年前からずっと言われていることなので、必ずしもノックアウトで機能が見つかるかっていうと、そうでもないと思うんですよね。ちゃんと適切に動いていくタイミングが分かってないと。
福永:そうですね。だから、ノックアウトで分かってないといっても、ストレスを与えたときにどうなるのかとかいう、そこまでいろいろ環境条件を振れば、また変わってくるというのもありますので。
尾崎:2018年のPLOS Biologyの論文で、GWASとかCRISPRとかsiRNAでのスクリーニングの結果から、あんまり論文書かれていないけど、機能がわかりそうな遺伝子は結構あるよという解析がありましたね。今はそこから4年たっているから、また状況は違うかもしれませんが、大規模にスクリーニングしても、誰も研究しない遺伝子は結構転がっているのかなという印象があります。
荒川:大腸菌のY genesがまずそうですよね。機能未知遺伝子ってやつですね。初期のゲノミクスで大腸菌でアノテーションが付かなかったものがY genes、「Y」から始まる遺伝子名が付いていて、当時3割ぐらいあって、この20年間で多分1割5分から2割ぐらいまでは減ったんですけど、いまだに機能が分かってないものも多くて。ただ、結構コンディショナルに重要な役割をするやつらは多いんですよ。そういうコンディショナルな状況っていうのを実験室内でなかなかつくってなかったから、そもそも発現もしなかったり、相互作用パートナーがいなかったりとかで、よく機能が分からない。いろんな変動をかけたところで、きちんとよく分かってないっていう状況が多かったんですけど。だから、まだしょせんその程度ですね。大腸菌の全ての遺伝子の機能すら全然分かってないっていう相当未熟なレベルに我々はいると思います。
岩崎:やっぱり10年前は、機能未知遺伝子の推定についてはもうちょっと10年後進んでてほしかったなって思っていた気もします。でも、あんまり本質的には変わってないな、そんな感じがします。
福永:それは結局、岩崎先生の言うところのロースループットな実験が必要になるって部分が解決されてないからっていうことでしょうか。
岩崎:そういうことですし、さっきのAlphaFoldの話もちょっと出ましたけど、配列を読み解くっていうことは、なかなか出来ていない。つまり、遺伝暗号の解読が50年前で、1960年代とかじゃないですか。そこから50年たっても、タンパク質のアミノ酸配列は分かるけど、その先が進まないっていう状況が50年間あまり変わらなかったのかなとは思います。
荒川:10年なんて一瞬ですから。
定量生物学的視点から10年を振り返る
岩崎:次は定量生物学的な観点を小林さんに伺うのはどうでしょうか。この10年を振り返っていかがでしょうか。
小林:まず10年前と比べて大きな変化だったなと思うのは、バイオインフォと定量の接点が大きくなってきたことですね(図3〜5)。定量計測とか理論は元々イメージジングと相性がいいんですけど、シーケンスやバイオインフォの間ではやりたいことがちょっとずれてて、これまで接点が少なかったと思います。ただ最近は、バイオインフォのほうからシングルセルRNA-seqの技術が出てきてから、分類とか同定だけでなく分化系譜のような、擬似時間的なものを推定しましょうという話が出てきています。


さらに、RNA velocityのような話題も出てきましたよね。スプライシングのモデルを入れて、遺伝子発現の時間的な変化方向も推定しましょうというふうになってきたことで、もともと時間変化やダイナミクスと理論とを組み合わせるといった研究は、どちらかというと定量のほうの話だったわけなんですけど、シーケンスのほうがこういう問題に結構近づいたというか、接点が大きくなってきて、いつの間にかワディントン・ランドスケープとかベクトル場のような話とかもバイオインフォからいろいろ出てくるようになってきています。 一方で、定量のほうは、10年ですごい進んだかっていうとそうでもないんだけど、順当に1個の細胞とかを測っていたところから、マイクロ流路とか、画像の自動追跡とかを使って、かなり多数の細胞を同時計測して、時間方向で見るみたいなことができるようになりました。だから、昔はシーケンスのほうが多数の細胞を見れて、イメージングは少数しか見れなかったという状況だったのが、定量の方も多数・多次元になってきている。最近だとラマンとか使って、イメージングなんだけどオミクス的に高次元情報も取ってきましょうみたいなの技術が出てきてるので、オーバーラップがかなり大きくなってきたなというのが、この10年で僕がとても感じる変化と思ってます。自分は定量の中でも理論系なので、実験のほうの人からすると、自分とはあまり接点ないよみたいな人もいるとは思うんですけど、理論系としてはやることがすごい広がって、10年たってより楽しくなってきたなという感じがあります。
岩崎:予想どおりという感じなんですかね。予想以上ですか?
小林:予想より早い感じですね。もうちょっとかかるかなとか思っていたんですけど、結構早いなと。最近RNA velocityのことを調べたりもしているんですが、使っている技術とか中のモデルとかを見ると、これが出てきてから、さらに近づいたと個人的には感じてます。
尾崎:最近、空間トランスクリプトームデータとかが取れるようになってきて、シーケンスベースの手法もあるんですけど、イメージングベースでマルチプレックスされたFISHみたいなデータを取って、それをシングルセルみたいに扱うという手法もあります。結構データ解析手法の論文とか出てきてるんですけど、イメージングで使われてるようなアルゴリズムを使ってたりとかもするので、データ解析手法も交流というか、混ざってきてるなって思って面白いです。
NGSとシングルセル解析
岩崎:シングルセル解析って、10年前はどんな感じでしたっけ。
尾崎:プレートベースの手法で、96ウェルに一個一個、細胞が入って、だからN=100もないという状況でした。それで、たくさん細胞があってすごい、みたいな。今は、数万とか数十万とか細胞を集めてきて解析するというような話になるので、もうすごいスピードで変わりましたね。
岩崎:シングルセルとか、空間トランスクリプトームみたいなのは、この数年でものすごい進みましたよね。そういうのは、案外みんな予想してなかったのか、みんな思ってたのか……。
荒川:Drop-seqありませんでしたっけ? 2012年の段階で。SMARTerは普通に使ってた気がするんですけど。
尾崎:Drop-seqは、2015年ぐらいですね。その前のプレートベースの技術は多分10年前にあったのかな。Tang et al. が確か2009年なので。
岩崎:だから、そういう分野ってのは、多分10年前に思ってたよりも進んだっていう、答え合わせ的な意味で言えば、図5図3図4そういうとこはありますか。
あとラマン光学もすごく進みましたよね。一方で、さっき言ったような非モデル生物のゲノミクスは、同じようなことをずっとやってるような気が私はしています。だから、そういう意味では、予想というか、期待どおりにはいかなかったかなっていう感じですかね。
福永:10年前は、BGIが「ゲノムをバシバシたくさん読むんだ」という雰囲気の時代だったような気がしますね。
尾崎:あとENCODEプロジェクトとかもありましたよね。
荒川:ナノポアとかで本当に気軽にみんながゲノム決められるようになったのは、予想できた未来だったけど、ちゃんと順当に民主化はされてきたなという気がしますね。
福永:10年前だとロングリードはPacBioは既にありましたね。
荒川:ナノポアがちゃんと製品を出したのは2016年からだと思うので、10年前はプロトタイプがやっと世の中に知られたぐらいの時期だったと思いますね。だから、本当に全てのラボでちゃんとゲノム読めるようなレベルになるっていうのは、簡単に予測ができた未来だったけども、ちゃんと実現してよかったなとは思います。
岩崎:あとは、配列類似性検索でDIAMONDとかMMseqs2のようなソフトウェアがこれだけ一般的に使われるようになるっていうのは、ちょっと予想してなかったですね。BLASTでみんなやってましたし、NCBIや配列アルゴリズムやソフトウェアを作るのが得意な人もたくさん取り組んでいた中で、こういう変化は正直予想していなかったです。
荒川:いや、速いのはずっと昔からたくさんあったんですけど、結局BLASTのほうが引用数もあるし、みんな使ってるし、レビュアーから文句言われないからっていう理由でBLASTが使われてるだけで、結構、内側ではみんな色んなツールを使ってたと思いますね。
尾崎:それで言うと、バイオインフォの論文って結局、一つのタスクにたくさんあって、でも、使われるのは少数っていうのは変わってないなっていうのは何となく思っています。ちょうど数日前に……。
岩崎:論文のジニ係数が高い、みたいなことが話題になっていましたね。
尾崎:そう。それを読んで、バイオインフォは知識積み上げ型の分子生物学と違うなと強く思いました。
岩崎:細胞生物学で流行ったのは、相分離とかですか、この10年。というか、最近ですけど。
小林:クロマチンとかもかなりやってますよね。
尾崎:そうですね。エピゲノムは、やっぱ読めるようになると、みんな測りたくなるっていうのはありますね。測ると、何か出てきたり、出てこなかったりする。
荒川:挑戦的なことを言えば、そういう技術の中でシングルセルって、いまいちできることに対して見つかったことが少ないというか、あんまりきちんと有効活用できてない印象なんですけど、その辺、尾崎さん、コメントあります?
尾崎:なんとなくですが、人間からデータ解析を離したほうがいいんじゃないかなっていうのはありますね。いろんな分野で使える技術なので、いろんな分野のシングルセルの話を聞いて思っているのは、基本的にデータ解析する人は、その同じ分野のシングルセル解析で、何が使われてるかっていうのを見て解析するんですけど、一方で、解析手法のカタログによって分かることって、もっとあるだろうと。そのデータで分かることはもっとあるはずなのに、みんなそうしないで論文書いて、論文書いたらもうシングルセルのデータはそのままみたいな感じになっている印象ですね。
荒川:そういう技術的にもいろいろと不幸な面があって、その空間情報とか細胞の種類っていう別のラベル情報がない状況でシングルセルやっても、よほどしっかり目的が固まっていないと、いい情報が取り出せないっていう中で技術が発展してきていますよね。すごく技術先行で、アプリケーションとしては取りあえず使ってみるみたいなものが結構多い時代があった部分は否めなくて。そういう意味では、今、ようやくそこのラベリングができるようになってきつつあるのかな。まだだいぶ不安はたくさんありますけれどもっていう状況かなと思います。
尾崎:それはおっしゃるとおりで、やっぱりうまく使えてる人って、特定の細胞を特異的に分取するための仕掛けを持ってる人とか、もともと細胞の分離のノウハウを持ってる人とかだったりするので、その細胞集団をうまく切り分けられないと、今のところうまく使いこなせてないかなっていう印象はあります。
荒川:新しいテクノロジーが論文レベルで出てくるのがまずあって、そこからそれが一般的な試薬化とか機械化されて販売されるまでに、5年から10年弱のタイムスパンがあって、さらに、そこからそれがユーザーが増えるのに、5年から10年ぐらいのスパンがありますよね。例えば、NGSの一番根本的な技術って、90年代半ばぐらいにはもう完全にできてたんだけど、2005年ぐらいに市販化されるまで、そんなに多くの人の手には渡ってなくて。でも、ちゃんとそれをみんな使い始めたのって、2010年とか、遅い人は2015年ぐらいからみたいな感じだったと思うんで、10年って、そういう意味では、テクノロジーができてから普及フェーズに入って、さらにユーザーが使って、集団でコミュニケーションができるまでっていうところの半分ぐらいの時間でしかないんですよね。
ちゃんと前が見えてる人にとってみると、10年前にはもうそのテクノロジーは論文としては存在しているので、それが世に広がることっていうのは目に見えている状況なはずなので、それが多分、ここで話が出ている、意外と予想どおりというか、あるいは何だったら、ちょっと期待外れぐらいなところに話が落ち着きがちな理由かなと思いますね。
生命システムの理解と創薬
岩崎:2000年ぐらいの『日経サイエンス』では、たとえばどの遺伝子をどの順番で処理すればアポトーシスへ導けるか、みたいなことがこれからシステム生物学で可能になる、と議論されていましたね。こういうことは言われていたように可能になってきたんでしょうか。
小林:どうですかね。あまり詳しく追ってないですけれども、そもそも何を操作すればそうなれるのかという問題と、操作できるのかという技術的な問題の二つがあると思います。その辺で言うと、合成生物学はシステム生物学と同時期ぐらい、2000年ぐらいにいっぱい出てきて一時期すごい流行りましたけど、2010年ぐらいにはちょっと下火だった気もします。でも、最近でも合成生物学って海外だと継続的にいろいろやられていて、思ったより息が長いなと思っていますね。カウフマンが描いたような工学よりの合成生物学は廃れちゃうかなと思ってたんです。実際、日本ではあまりやられていなくて、むしろ、プロトセルとか細胞を創る会とかのアクティビティのほうが高くなっいたんですけ。カウフマンっぽい考え方のやつは、海外ではまだ生き残ってて、ちゃんと積み上げて成果が出てきてるなというのはあります。
岩崎:あと創薬ですけど、創薬も、バイオインフォマティクスは10年前よりは確実に貢献していると思うんですけど、昔考えられていたようなバラ色の未来というほどでは、やっぱりないかなと。着実に進んではいるけど、そんなに理想的には進んでない。
荒川:mRNAワクチンは、完全にバイオインフォマティクスじゃないですか。
岩崎:それは、ちょっと私は何とも言えません。もうちょっと地道な生化学という感じがしてましたけど。
荒川:RNAのアジュバントだったり、アジュバント効果を抑制したりというようなところはもちろん生化学ですけども、そのターゲットを決めて簡単に設計できるっていうところで。
岩崎:そうですけど、「あれはバイオインフォマティクスの成果です」とか言ったら、ちょっと怒られそうじゃないかな。
荒川:もちろん。でも、最も理想的な設計が作用できるドメインに落とし込んでるなということですね。
岩崎:疾患遺伝子の研究とかも、ヒトの個人ゲノム解析がものすごい進んだので、いろんな発見はありましたよね。だけど、遺伝率だけでは説明できないものがどうしても残った。いろんな分野ですごい進んだけど、やっぱり生体分子とか生命の複雑さというものの本質は、まだつかめてないかなって。勝手にまとめてますけど。
10年前にも話したことですけど、配列相同性検索によって基本の分子生物学知識と結びつけていくのは、このペースで配列が来ると計算が大変だし、しんどい。よく中村保一さんも言っていたけど、予想の予想の予想の予想みたいになっちゃうので、怪しい結果だし、結局どうするかは実験をしないといけない。計算コストがかかりすぎるので、分子計算のようなことがいいのではということを言っていました。分子計算という感じにはならなかったけど、結局GPUとかでまた違った計算が可能になったかなというふうには思ってます。実験室進化みたいなのはできますけどね。あとやっぱり配列相同性検索は既存知識の枠を出ないので、ab initio解析できるように技を増やす。これは、やっぱりAlphaFoldのように技が増えたのかなと。
あと分子生物学をすっ飛ばす、とも10年前に言いましたが、これは、一回遺伝子の知識にしないで配列から直接表現型を予測するとか、メタゲノムデータから直接、環境診断するとか、そういうことをやったらいいんじゃないかなっていうことですね。この辺は、まだこれからっていうところなのかな。でも、こういうこともちょっとやられてますよね。そんな感じの10年でした。
創薬は難しくなってきている
尾崎:大上さん、創薬は楽になりましたか。
大上:厳しくなってるんですよね。10年前に思っていたことは、私、計算屋なので、10年たてばスパコンも進化するし、手元のノートパソコンだって速くなるし、できることは増えるよねっていうのは、もちろん考えていて(図6)。当時10年前から、もちろん機械学習、当時はAIって呼ばれ方をしてませんでしたけど、機械学習が技術的にはたくさんあったので、機械学習でバイオインフォマティクスの応用はたくさんあるだろうなって、これからも増えるだろうなっていうのは、本当に予想どおりだし、計算機がそのまんま速くなったのも予想どおりでした。使えるデータも、もちろんたくさん増えましたし、いろんなことができるようになりましたねというのは、変わらず予想どおりという感じですよね。ただ一方で、こういう実験のデータって全然ないよねとか、こういう疾病の薬のデータがないよねとか、そういう領域も相変わらず足りないですよねっていうのも現時点で変わってないですよねっていう感じです。

その中で、マイナーですけど結構変わったなと思ってるのは、思ったより機械学習が流行っててびっくりしてるっていうことと、AIの浸透度が尋常じゃないなっていうのがあります。あとは、その影響もあって、CPUで計算するんじゃなくて、用途特化型のハードウェア、要は、GPUとTensorProcessing Unitとか量子コンピューターとかって言いだしてる人たちがたくさんいるとか。あとはFPGAでもいい感じに仕立てたFPGAがあったりとか。本当にいろんなハードウェアが増えて、それをみんなが使いこなしてるのは、ちょっと想像を超えたところがあります。ただ、総じてあんまり変わってないですよね。
10年後にじゃあどうなるかっていうと、まず量が増えるっていうのは想像の範囲ですし、一方で何か質が変わるかっていうと、これは自分自信の課題でもありますけど、それはそんなに期待できないなっていうところはあります。ただし、量が増えた結果、新しいように見える、質的に変わったように見せられるような成果はいくつか出せるんじゃないかなと思っています。僕が興味あるのは創薬ですけど、でも一方でやっぱりあらゆるところがケース・バイ・ケースだし、パラメーターの組合せの沼にはまっていく未来がずっと続くだろうなというのは思っているところです。
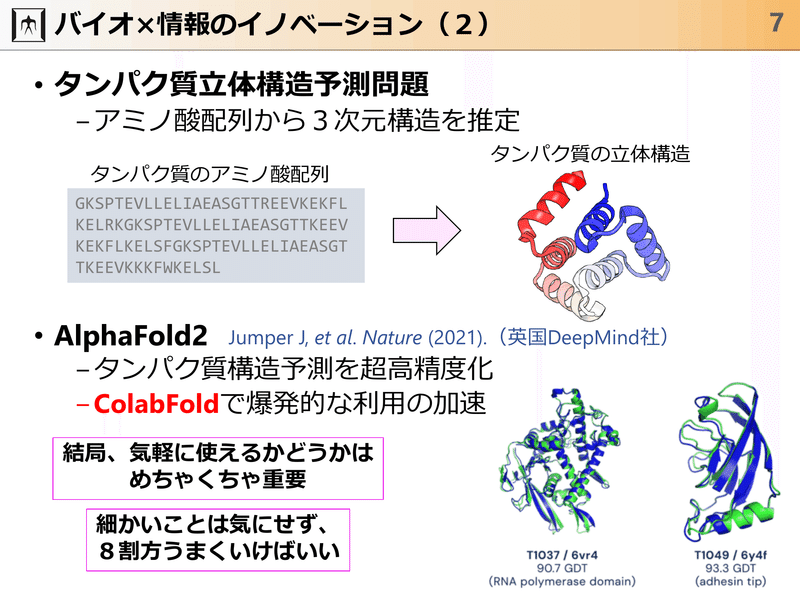
大きかったのはBLASTとAlphaFold2なのかなとは思っていて、AlphaFold2は想像を遥かに超えて爆発的に普及してるなというふうには感じています(図7〜8)。AlphaFold2の一連の出来事で、結局気軽に使えるかどうかっていうのがまず一番本当に重要で、しかも、細かいことはあんまり気にしなくてもいいんだなっていうのはちょっと感じました。実際にうまく予測できない配列だってたくさんあるんですけど、8割方うまくいけば、すげえってみんな言ってくれるっていうのはそのとおりだと思うので、細かいことを気にせずにうまくいくようなものを作れれば、それでいいんじゃないかっていうのは今後も多分続くんだろうなと思っています。

僕はPPI (タンパク質間相互作用)の予測っていう話をやってますけど、今でも実験的にどんどん新しいPPIが発見されていて、1年で10万ペアくらいは新しく見つかってるんですよね。じゃあ実際どのくらいありそうかっていうのも、これは妄想で試算すると、ヒトでもあと2.5倍ぐらいあってもいいんじゃないかと思います。要は、まだまだ実験的にPPIだって見つけることができるし、それらに何らかのアノテーションが付いてこれはとても重要な機能だっていうふうに言われる可能性だってあるし、それが創薬標的になるなんて話ももちろんあるということで、やれることはたくさんありそうだなというのは簡単に想像ができます。
スパコン「富岳」は話題ですが、今後このペースで計算機が発展するかどうかはちょっと分からりません。でもきっと伸び続けるだろうと思っています。スパコンって、大体10年で1,000倍ぐらいの計算の速度の向上が続いているので、このペースで10年後に1,000倍の計算能力が実現したら、目の前のパソコンだって1,000倍ぐらいの性能になってるかもしれない。「京」ぐらいの性能を持つノートPCになってるかもしれないなって、勝手に想像はしています。最近、MDシミュレーションとかでも、すごく大変な計算が何とか頑張ればできるようになってきたっていう事例が増えています。私たちがやってるのは細胞の膜を透過する分子の計算なんですけど、こういうシミュレーションができるようになってきてようやく、細胞の中に入りやすい分子をデザインするとか、PPI制御ができるペプチド配列を設計するとか、ヒトをコントロールするための手数が増えてきたかなという感じはしています。1,000倍の速さになったときに何ができるかなと、よく考えています。あんまり答えはないですが。
岩崎:立体構造のペアってのは、もう全部、2、3年後には分かっちゃうんじゃないんですか。
大上:予測では分かってるんじゃないですかね、きっと。
岩崎:もうタンパク質配列を与えれば、結合する小分子も2、3年ぐらいしたら、結構予測できるようになるんじゃないかなと思ってるんですけど。だから、機能未知遺伝子も構造を予測して、何と結合するかみたいなことは、2、3年後ぐらいには、もっとハイスループットに分かってもおかしくないなと。甘いですか。
大上:低分子に関しては、多分、この標的に結合するっぽいといった情報を列挙することは今でもできると思います。ただ、それを列挙したところで、その後の開発の部分で、これは難しい、これはちょっと物性が、とかっていう話になってくるので、困難はまだまだたくさんあるんですけど。
岩崎:あとは転写因子を入れたら、結合DNA配列が分かっちゃうとかもできそうですよね。 そうすると、その先に、システムの青写真みたいなのが書けると思うんですけど、生命システムをコンピューター上で再現するための。その先は、どうなるのかな。
液-液相分離のインパクト
尾崎:例えば、今、相分離(Liquid–liquid phase separation;LLPS)ってバイオインフォでやってる人っています? 最近ではデータベースをつくってる人とか相分離するタンパク質をコードする遺伝子を予測するとかがありますよね。
大上:分子シミュレーション界隈では、結構やってるんですよね。
荒川:どういう部位が相分離に効くかとか、相分離のときのパートナーが何になるかとか、そういうようなところっていうのは、やってますね。
岩崎:アルギニンがどうとか、芳香族アミノ酸残基が大事とか、あとシンプルリピートが大事だとか、いろんな人がいろんなことを言っていて、私もよく分かってないんですけど。でも、細胞内コンパートメントとかを扱おうと思ったら、どうするんですか。やっぱりミカエリス・メンテンとかじゃ駄目ですよね。
荒川:そこ、まさに僕がこの10年間で最も意外だったというか、大きな発展だったところで、今まで分子クラウディングって言ってたところとか、in vitroとin vivoでのkineticsの違いみたいにいってたところが、LLPSで大体全部説明できてしまうので、最近あまりクラウディングって言わなくなりましたよね。そこが分かってきたおかげで、いろんなことが統一的に考えられるようになってきたかなというのが、ここ5年間ぐらいかなと思いますけど、僕的に一番意外だったことですかね。
小林:これ、でも、結局in vitroとin vivoで速度定数のスケールが合わないみたいな話なのか、それとも、岩崎さんが言われてたみたいに、ミカエリス・メンテンでは駄目という話なのか。後者なら、反応速度の関数形というか、反応系のキネティクスの法則自体をもうちょっと考えなきゃいけないと思うんですけど、どうなんですかね。相分離のほうで新しい反応をうまく表現するための新しい式みたいなのができてるってことですか。それとも、パラメーターの整合性がうまくとれるようになったというか、一応説明できるようになったということなのか?
荒川:要するに、状態によって物質の濃度を一定のコンパートメントに高濃度で取り込み続けられるし、あるタンパクからあるタンパクへの受け渡しってところが、ものすごく狭い空間スケールの中で行えるということになるので、基質の量とKmの値のところっていうのが、コンパートメント依存で変わっちゃうっていうことですよね。だから、ミカエリス・メンテンの延長で説明できなくはないんだけど、もはやミカエリス・メンテンで考える必要があんまりないというか、ミカエリス・メンテン自体を変数に入れた、もう1次上の式がないと、ちゃんと表現し切れない。
それをやるぐらいだったら、もはや最適化を入れちゃったような計算式でも、ある意味、きちんと動いてしまうというとこですね。だから、意外とSシステムとかでやってたことってのが、そんなに間違ってなくて、最適化するっていう条件を入れてしまうと、実は生物の中でも、各反応が、そういうコンパートメンテーションレベルで最適化されていたので、実は正しい挙動をやっていたっていう。だから、ミカエリス・メンテンで一個一個作るよりも、むしろ、そういうほうが自然に当てはまっていたっていう。代謝で言えば、そういう感じですかね。代謝以外のところは、なかなかそれが当てはまらないところがありますけど。
尾崎:そしたら、システム生物学も、そういうLLPSを考慮に入れると、今までよりも予測が当たるようになるっていうことですか。
荒川:システム生物学ってそもそも学問じゃないと思うんで、あれが何かをやってたかよく分かんないんですけど、かなりLLPSを入れることで説明がつくようになったことはたくさんあって、結局、僕、クマムシもクモの糸も両方とも、最終的にLLPSに行き着いちゃってるっていう状況です。
ゲノムの解読に必要な情報
尾崎:福永さんは、10年前の予想はどうでしたか。
福永:まだ10年前は、正直10年後のことはあまり深く考えられてなかったですね。荒川さんと岩崎さんの原稿は、非常に勉強になり、何度も読み返していましたが。
あと、荒川さんが講演で、Debora Marksのタンパク質立体構造予測の論文を非常に押していて、今後こういうことをやらなきゃいけないんだということを主張していたのは、すごい印象に残ってます。
荒川:これ、1本だけ引用してて(図9)、予想どおりAlphaFoldは、この方向で来たなっていうところで。僕、この発表は実は2007年ぐらいから聞いてて。ずっと論文出ないなと思いながら待ってて、やっとここで出たっていう感じなので、もうこれが発表されたときには、かなり私は完全に確信になってたんですけど、多分初めて聞くと、なかなかイメージしにくいところもあったのかなと。

ゲノムの解読をするっていう、さっき岩崎さんがおっしゃった、まさにそこを本当に攻めなきゃいけないところで、それを全然攻め切れていなかったけど、もっとまじめにやりましょうと。やったらAlphaFoldみたいなことになるよってことだと思います。今、うちのラボでは、hypothetical遺伝子って名前を使うことを禁止していますが、それでちゃんと議論が成立しています。やっぱり何かしら構造とか、そういうとこからエビデンスを持ってきて、せめて何をやってるか、何やってそうか、どんなときに動いてて、こう思われるかっていうところを議論しないで、例えば「GOだけで全体のプロットとか出すのは、まじやめろ」っていうようなことを言うようなレベルにまで来たので、そこは10年間で非常に大きな変化だったかなと思いますね。
やっぱそれができるのは、ゲノムっていうのは独立情報なので、それをベースに学問が構築できるというのが非常に大きいので、われわれは、これをやっぱり誇りに思ったほうがいいし、それをより継続的にいろんなことをやったほうがいいだろうなというふうに考えています。僕が結局この10年間でやってきたのは、その部分もあります。クモ糸で配列と物性のデータ、たくさん取って、1塩基レベルでどういう物性が変わるかっていうのを一応、予測できるようになって、実際にそれでいい製品を作れるようになったりとか、そういったこともできるようになってきているので、ゲノム情報から表現型をちゃんと定量的に予測ができるようできるようになってきたかなと思うところですね(図10)。

結局、その遺伝子がいつどこで発現するか、どれぐらい発現するか、何と相互作用するか、何をリガンドにするかっていうことは全部ゲノムに書いてあるはずなんで、書いてあることは、僕らは解読しなきゃいけなくて、できるはずなんですよ。だから、それを本気でやらなきゃいけなくて、できないとか、この情報が足りないとか、この技術が足りないって思うんじゃなくて、やるっていう気持ちで、ちゃんとやっていかなきゃいけないなということを、ずっと10年前から言っていて思っているという感じですね。同じようなことが、今後10年間もどんどん起きてくるでしょう。だから、やっとタンパクがどうフォールドするかが分かったので、これから何とインタラクトするかとか、何をリガンドにするかとか、どう発現制御をするとかとか、そういうようなことっていうのが、かなり分かってくるだろうなと思いますね。
尾崎:とはいえ、データが足りないと思うんですよね。
荒川:いや、進化の情報とか入れると、結構情報がありますし、データがなければ取ればいいだけなんですよね。データ取るっていうときに、トランスクリプトームとかって、どういう条件で取るかのほうが重要だから、データベースにしても、あんま意味がなくて、実験条件も違えば、取ったタイミングも、厳密には、ちょっと違うようなデータが山ほどデータベースにあっても、そこから何もマイニングできないんですよ。だから、こういうデータが取りたいっていうふうに思って、何個データが必要だっていうことを計画して、そこで1,000個でも1万個でも取ればよくて、今、別に1,000個、1万個のトランスクリプトームなんて余裕で取れるじゃないですか。だから、そのデザインこそが重要で、データがあるかないかは、今は多分もう、あんまり重要じゃないんじゃないかなと思いますね。
尾崎:先ほど大腸菌でコンディショナルな状況を振るのが大事ってお話があったと思うんですけど、大腸菌でも、あとは多細胞生物だと、セルタイプとか、そういうコンディション振っていくとか、あとは細胞の空間のコンテキストを振っていくというのが大事だと思うんですけど。実験系の人と話すと、どうしてもやりやすいコンディションと、やりづらいコンディションみたいなのがあって、そこがまだ自由度が低いなっていうのが、すごいありますね(図11)。この細胞だったらいいけど、こっちの細胞は実験系をゼロから作り始めるのが時間かかるっていうのがあって。でも、やっぱりそういうコンディショナルなところを詰めていかないと、先ほどのようなゲノムに書いてあることって解読できないんじゃないかなっていうのがあって。それは結局、10年前は、もう解決してると思ってたんですけど、あんまり解決してなくて。

荒川:結局、だから、その条件を探してやろうって気になってないですよね。だから、そういう細胞モデルが必要だってなったときに、じゃあ、こういう動かし方をしたら、そういう細胞系になるっていうことを予測して、実験系も組めるようになってくると、どういうデータポイントを取るかっていうことにつながってくるかなと思います。
福永:尾崎さんが言うデータが必要っていうのは、具体的に、例えば、どの遺伝子の機能を解き明かしたいとかいうときに、どういうデータが必要なんですか。ちょっといまいちぴんとこなかったんですけど。データが足りないっていうのは、もちろん、例えば、活性のデータとかは全然足りないと思うんですけど、動態のデータとか、発現周りに関しては、データが足りないっていうのは、具体的にどういうことですか。
尾崎:時系列データが足りないっていうのが、まず一つあります。時系列が稠密になれば、前後関係が分かって、どっちが、因果関係が分かるのに、取りづらいから取れないって。取りづらいから取ってないっていう。もちろん擬時間解析とかはあるんですけど。
福永:でも、さっきの荒川さんのロジックに従えば、もちろん時系列データは、いわゆるカンニングのために使えるとは思いますけど、本質的にゲノムに書いてあるって考えれば、時系列データを使わなくても本当は解けたりするんですか。解けたりするものを、点を上げるために時系列データを使いたいって話なんですか。 もちろんデータがいっぱいあるに越したことはないと思うんですけど、荒川さんの発言を聞いてたら、ちょっと多分、大多数の人は本気度が足りないんだなっていうのを何となく思いました。
尾崎:確かに。ちょっと自分でも、データを与えられることに慣れ過ぎているなという反省があります。だから、「取りあえずもっとたくさんデータがあったら、もっといろんなことが分かるだろう」ということをナイーブに信じているなっていうのを少し反省しました。なので、荒川さんが言ってたことにつながると思うんですけど、当たり前のことですが、バイオインフォの側からこういうデータを取りましょうっていうのをもっと言わないといけないかなと。そのときに、もう少し実験系の人が思い付かない範囲で、そういうことが言えないといけないんだろうなというのは思います。
荒川:僕らが主導権を握って実験計画からやっていく時代になるわけですよ。10年後の話をすると、ゲノムwritingが確実に普及しますよね。長いDNAを簡単に安く書けるようになってくる。今まで読めただけなんだけど。そうすると、どういうふうにそれを設計しようかっていうところがキーポイントになってきて、そんなの多分、もう本当に真のバイオインフォマティシャンじゃないとできないんですよね。だから、そこの実験計画から本当に僕らが主導するサイエンスをつくると、かっこいいんじゃないですかね。
(次号、後編に続く)
本記事は日本バイオインフォマティクス学会ニュースレター第41号(2022年3月発行)に掲載されたものです。
以下のURLにて、全ての記事を無料でお読みいただけます。https://www.jsbi.org/publication/newsletter/
この記事が気に入ったらサポートをしてみませんか?