SARS-CoV-2 スパイクはインテグリンとTGF-βシグナルを介してバリア機能障害と血管漏出を誘発する

要旨
重症のCOVID-19は、肺だけでなく遠位臓器における上皮および内皮のバリア機能障害と関連している。炎症反応の亢進がバリア機能障害と関連していることは理解されているが、血管漏出の引き金については不明である。我々は、SARS-CoV-2のスパイク(S)糖タンパク質と上皮・内皮細胞との細胞内相互作用が、ウイルスの複製やACE2受容体とは独立して、in vitroでのバリア機能障害やvivoでの血管漏出を引き起こすのに十分であることを報告する。我々は、細胞外マトリックスの再編成とTGF-βシグナルに関連するS-トリガー転写反応を同定した。遺伝子ノックアウトと特異的阻害剤を用いて、グリコサミノグリカン、インテグリン、TGF-βシグナル伝達軸がSによるバリア機能障害に必要であることを証明した。特に、SARS-CoV-2感染により生体内で漏出が起こり、インテグリンを阻害することにより漏出が抑制されることを示した。この結果は、SARS-CoV-2感染によって引き起こされる血管漏出の機構的洞察をもたらし、COVID-19を標的とした治療法開発の出発点となるものである。
はじめに
重症急性呼吸器症候群コロナウイルス-2(SARS-CoV-2)はコロナウイルス科に属するヒト病原体で、コロナウイルス病2019(COVID-19)の原因ウイルスである。SARS-CoV-2感染の転帰は、無症状からインフルエンザ様症状を伴う非重症のCOVID-19まであり、急性肺障害や急性呼吸窮迫症候群(ARDS)を伴う重症例に進行することがあります1,2,3。重症COVID-19の肺病理は、炎症反応の増悪によって誘発されると考えられる上皮および内皮のバリア機能障害に起因する肺水腫を含みますが、上皮・内皮の過透過性の具体的誘因および特定のウイルス因子の関与は十分に解明されていません。
SARS-CoV-2は、約30kbのポジティブセンスRNAゲノムを持ち、スパイク(S)、ヌクレオキャプシド(N)、マトリックス(M)、エンベロープ(E)の4つの構造タンパク質を含む約29種類のタンパク質をコードしています7,8。S糖タンパク質のホモトリマーはSARS-CoV-2ウイルスを被覆し、感受性細胞の表面にあるウイルス受容体、アンジオテンシン変換酵素2 (ACE2) と結合してウイルス侵入を媒介する9,10.Sは、ACE2と結合する受容体結合ドメイン(RBD)を含むS1と、ウイルスと細胞膜の融合に必要な融合機構を含むS2の2つのサブユニットから構成されている7,11,12。S1とS2は、S1/S2およびS2'という2つの切断部位で隔てられており、Sがウイルスと細胞の融合を媒介するためには、宿主プロテアーゼによって切断される必要がある。Furin-like proteases, cathepsin L, TMPRSS2はこれらの部位を切断することができ、SARS-CoV-2感染に必須の宿主因子となっている10,13,14,15.ACE2がRBDと結合すると、Sの構造変化が起こり、S1が脱落し、融合ペプチドが宿主膜に挿入される16,17。
SARS-CoV-2のS糖タンパク質は、ACE2に加え、ヘパラン硫酸含有プロテオグリカン(HSPG)やインテグリンを含む多くの細胞表面因子と結合することが報告されており、これらはSARS-CoV-2の侵入を促す付着因子として機能することが提案されている18、19、20。ウイルスの侵入を促進するだけでなく、Sがこれらの宿主因子と結合することで、肺の病態に寄与するシグナル伝達経路を媒介する可能性がある。実際、SARS-CoV-1のSがACE2に結合すると、細胞表面からACE2が枯渇し、レニン-アンジオテンシン系のバランスが崩れ、炎症反応、バリア機能障害、肺損傷を促進することが証明された21,22,23。SARS-CoV-2 S24,25,26,27,28 についても、同様のACE2依存性の経路が報告されている。SARS-CoV-2の侵入カスケードのユニークな要素は、RBDを含むSのS1部分がACE2レセプターとの結合後にビリオンの表面から排出されることであり、排出されたS1がビリオンとは別に上皮細胞や内皮細胞と相互作用することが示唆されている16,17。SARS-CoV-2のS糖タンパク質と細胞表面の相互作用はバリア機能障害を促進すると考えられるが、そのメカニズムや関与する宿主因子は十分に解明されてはいない。
我々や他の研究者は、フラビウイルス非構造タンパク質1 (NS1) などのウイルスタンパク質が内皮細胞と相互作用し、内皮グリコカリックス層 (EGL) や細胞間結合複合体など内皮バリアの完全性に必要な細胞構造の破壊を仲介するシグナルカスケードを引き起こす現象について述べている29,30,31,32,33,34.ここでは、SARS-CoV-2 Sがin vitroでの内皮・上皮バリア機能障害およびin vivoでの血管漏出に寄与しているかどうかを検討した。その結果、SARS-CoV-2の全長SとRBDは、ACE2非依存的にバリア機能障害と血管漏出を引き起こすのに十分であることが明らかとなった。さらに、転写解析により、Sが細胞外マトリックス(ECM)の制御に関わる転写産物の発現を調節することが示され、実験的検証により、グリコサミノグリカン(GAG)、インテグリン、トランスフォーミング成長因子β(TGF-β)シグナルがバリア機能障害に必要であるメカニズムが明らかにされた。最後に、SARS-CoV-2の生体内感染がマウスの肺の血管漏出を引き起こし、それがインテグリンの拮抗によって回復することを見いだした。これらのデータは、バリア機能障害を促進するSARS-CoV-2 Sの役割を明らかにし、このプロセスに対する重要なメカニズム的洞察を与えるものである。
研究成果
SARS-CoV-2 Sは内皮および上皮の透過性亢進を媒介する
SARS-CoV-2 SがSARS-CoV-2感染とは無関係にバリア機能障害を引き起こすかどうかを調べるために、我々は経上皮/内皮電気抵抗(TEER)アッセイを利用した。TEERを用い、バリア透過性の代用として、トランスウェルの頂部チャンバーに播種したヒト肺微小血管内皮細胞(HPMEC)またはヒト肺上皮細胞(Caly-3)の単層間の電気抵抗を測定した(Fig. 1A)。HPMECを代表的な内皮細胞として選択したのは、COVID-19に関連する肺病理学的特徴に加え、ACE2を内因性に発現せず、SARS-CoV-2感染に対して寛容ではないため、ウイルス感染とSを介したバリア機能障害を分離できるためである(図S1A, B)。我々は、組換え可溶性三量体SとRBDを自社製造し、高純度であることを確認した(図S1C, D, G)

B DENV2 NS1(5μg/mL)、VEGF(50ng/mL)、SARS-CoV-2 S(10μg/mL)を含む指定処理でHPMEC単層のバリア機能を経時的に測定したTEERアッセイのタイムコース。データはn = 3生物学的複製から得たものである。
C 示した処理から24時間後のHPMECおよびHPMEC/ACE2の単層膜のバリア性を測定するTEERアッセイである。VEGF陽性対照(50 ng/mL)。データは、n = 4生物学的複製から得たものである。
D Cと同様であるが、指示されたVSV疑似型粒子で指示された希釈率で処理した。VSV-baldとVSV-Gは1:30で希釈。データは、n = 4生物学的複製から得たものである。
E Cと同じだが、指示された濃度のSARS-CoV-2 RBDで処理した。データはn = 3生物学的複製からのものである。
F Cと同じであるが、Calu-3細胞単層のバリアーを測定。データはn = 3生物学的複製からのものである。
G 抗S抗体のカクテルが、Sを介した内皮過透過性を阻害する能力を測定するTEER阻害アッセイ。S(10μg/mL)と抗体カクテル(各抗体15μg/mL;1A9 [Genetex] およびCR3022 [Absolute Antibody] )をトランスウェルインサートの上部チャンバーにHPMECまたはHPMEC/ACE2の単層に同時に加え、TEERは処置後24時間(hpt)に測定された。データはn = 3生物学的複製からのものである。すべてのパネルで、点線は未処理対照条件の正規化TEER値である。すべてのデータは、平均値+/- SEMでプロットされている。すべてのパネルにおいて、値はTukeyの多重比較検定付き一元配置ANOVAによって未処理対照と比較されている(B)以外は、両側不対t検定によって分析されている。 *p < 0.05, **p < 0.01, ***p < 0.001, および n.s. p > 0.05.ソースデータはSource Dataファイルとして提供される。
親細胞のHPMECを可溶性三量体SARS-CoV-2 Sで、また陽性対照としてデングウイルス(DENV)NS1および血管内皮増殖因子(VEGF)で処理した。先に見たように、HPMECをDENV NS1で処理すると、処理後6時間(hpt)をピークにバリア抵抗性が可逆的に減少した(Fig. 1B)。興味深いことに、SARS-CoV-2 Sも親細胞のHPMECに可逆的なバリア機能障害を引き起こしたが、その時間動態は異なり、約24時間後にピークに達した。HPMECはACE2を内因性に発現していないので、これらのデータは、SがACE2とは無関係にバリア機能障害を引き起こすことを示唆している(図1B)。Sによる内皮の透過性亢進は24時間後にピークに達することから、この時点をその後の実験に利用した。次に、親細胞のHPMECまたはヒトACE2を過剰発現させたHPMECを可溶性三量体SARS-CoV-2で処理したところ、Sは用量依存的に内皮の過透過性を引き起こすことがわかった(図1C)。このことは、この表現型がACE2とは無関係であることを示唆している(Fig. 1C)。ビリオンと結合したSが内皮の過透水性を引き起こすかどうかを調べるために、SARS-CoV-2 S(VSV-S)とVSV糖タンパク(VSV-G)または糖タンパクなし(VSV-bald)を発現したVSVをネガティブコントロールとして使用した。可溶性SやVEGF陽性対照と同様に、VSV-Sも用量依存的に内皮の過透過性を引き起こしたが、VSV-GとVSV-baldはバリア機能に影響を与えなかった。HPMECとHPMEC/ACE2の結果を比較すると、ここでも相対的なTEER値に有意差は認められなかった(図1D)。さらに、組換えSARS-CoV-2 S RBDは、用量依存的にHPMECのTEERの低下を引き起こすのに十分であることが分かった(Fig. 1E)。次に、TEERアッセイで上皮Calu-3細胞の透過性亢進を誘発するSの能力をテストし、全長三量体SとRBDが上皮の透過性亢進を誘発するのに十分であることを確かに見出した(Fig. 1F)。この表現型がSに特異的であることを確認するために、抗S抗体によるS誘導のバリア機能不全の抑制能力を測定したところ、2種類の抗S抗体のカクテルがSによる内皮の過透過性を消失させることがわかった(Fig. 1G)。SARS-CoV-2のSがHPMECの内皮過透過性を誘発する能力は、近縁のコロナウイルス間で保存されているかどうかを調べるために、高純度と判断したヒトコロナウイルス(HCoV)-229EおよびHCoV-OC43から完全長のSおよびRBDを作成した(図S1E-G)。興味深いことに、HCoV-229EおよびHCoV-OC43からのSおよびRBDは、SARS-CoV-2 Sとは対照的に、HPMECにおいて内皮の過透過性を引き起こすことができず、この特性がSARS-CoV-2 Sに特有であることを示唆した(図 S1H)。これらのデータは、SARS-CoV-2 SとそのRBDが、ヒト肺内皮細胞および上皮細胞の両方においてバリア過透過性を促進することを示唆している。
SARS-CoV-2 Sは内皮および上皮のグリコカリックス層を破壊するきっかけとなる
上皮および内皮表面の細胞表面タンパク質は、上皮/内皮グリコカリックス層(EGL)と呼ばれる糖鎖の密なメッシュによって取り囲まれている。EGLはシアル酸やグリコサミノグリカン(GAG)を含み、バリア機能の重要な決定要因として、上皮および内皮細胞をシアストレスから保護する役割を担っている35。我々の以前の研究により、フラビウイルスNS1糖タンパク質は、EGL分解酵素の活性化を介したEGLの破壊を通じて、内皮機能障害を媒介することが証明されている30,32。SARS-CoV-2 SがEGLを破壊するかどうかを調べるために、HPMECとCalu-3細胞をSで処理し、シアル酸(SIA)、ヘパラン硫酸(HS)、ヒアルロン酸(HA)、コンドロイチン硫酸(CS)などのEGL主要成分の表面レベルを免疫蛍光法(IFA)によって測定した。また、ヒアルロン酸を分解するヒアルロニダーゼとシアリル化糖鎖を分解するノイラミニダーゼのレベルも測定した。その結果、HPMECとCalu-3細胞の両方でS処理をすると、EGL成分がコントロール条件に比べて著しく減少し、逆にEGL分解酵素がアップレギュレートされることがわかった(Fig. 2A-H)。これらのデータは、フラビウイルスNS1と同様に、SARS-CoV-2 SがEGLの破壊を媒介することを示唆している。

B Aと同様であるが、細胞を透過処理してから、指定されたEGL破壊酵素の染色を行った。n = 3生物学的複製からの代表的な画像を表示します。
C n = 3生物学的複製からのAの定量化。
D n = 3生物学的複製からのBの定量化。
E Aと同様であるが、Calu-3細胞単層のEGL破壊を測定した。n = 3 の生物学的複製からの代表画像を表示。
F Bと同様であるが、Calu-3細胞におけるEGL破壊酵素の発現を測定した。n = 3生物学的複製からの代表的な画像を表示します。
G n = 3 biological replicatesから得たEの定量。H n = 3 biological replicatesから得たFの定量化。すべての画像において、核は青色でHoechst、表示された糖鎖は緑色でプローブされ、スケールバーは50 µmであった。点線は正規化した無処理対照条件。MFIは平均蛍光強度である。すべてのデータは平均値+/- SEMでプロットされ、未処理対照と比較して、両側不対t検定により*p < 0.05, **p < 0.01, ***p < 0.001, n.s. p > 0.05 とした。ソースデータはSource Dataファイルとして提供される。
しかし、細胞表面に存在する低レベルのACE2が我々の表現型に寄与している可能性を排除するために、CRISPR-Cas9を用いてACE2遺伝子に二本鎖切断を導入した。Sを介したEGL破壊は、ACE2標的ガイドRNAをコードするレンチウイルスまたは非標的ガイド(NT)を導入したHPMECで同等であったことから、SARS-CoV-2のSをトリガーとするバリア機能障害は、ACE2とは独立して起こりうることがさらに裏付けられた(図S2A、図B)。最後に、懸念される複数のSARS-CoV-2変異体由来のSがバリア機能障害を引き起こすかどうかを検証したところ、祖先のWuhan変異体由来のSタンパク質と同様に、α、β、γ、δおよびomicon変異体由来のSはすべてHPMEC上のEGL破壊を促進できることがわかった。この現象は、1つの変異体に特有のものではなく、SARS-CoV-2のSに一般化できる性質であることがわかる (Fig. S2C, D).最後に、SARS-CoV-2ウイルスがEGL破壊を促進するかどうかを調べるために、HPMECにSまたはSARS-CoV-2ライブウイルスをMOI 5で接種した。その結果、組換えSとウイルス粒子の両方がEGLの破壊を引き起こすのに十分であり、遊離Sとウイルスに結合したSの両方が内皮機能障害を引き起こす可能性があることをさらに証明した(図S2E、F)。
SARS-CoV-2のSとウイルス感染がin vivoでの血管漏出を引き起こす
SARS-CoV-2 Sがin vivoで血管漏出を引き起こすかどうかを調べるために、我々が以前に開発した皮膚漏出モデルを用いた。このモデルでは、マウスの背側真皮の異なるスポットに化合物を局所的に皮内投与し、トレーサーとしてAlexa fluor 680と結合したデキストランを静脈内投与した。2時間の処置後、マウスを安楽死させ、背部真皮におけるdextran-680の局所相対集積を蛍光スキャナーで測定した31,33,36。SARS-CoV-2 SがACE2非依存的にバリア機能障害を媒介するという我々の観察を踏まえ、我々はヒトACE2を発現せず、武漢やワシントン分離株を含むほとんどのSARS-CoV-2変種による感染に寛容でないWT C57BL/6 Jマウスを使用した9、37.我々は、DENV NS1陽性対照と比較して、SARS-CoV-2 Sが対照条件よりもマウスの背側真皮に血管漏出を誘発することを観察し、SARS-CoV-2 Sが生体内で血管漏出を引き起こすのに十分であることを示した(図3A、B)。SARS-CoV-2 Sがより生理的に適切な経路で投与された場合に血管漏出を媒介するかどうかを調べるため、Sを経鼻投与し、様々な臓器におけるdextran-680の蓄積量を測定して局所(肺)および遠位(脾臓、小腸、肝臓、脳)両方の血管漏出量を評価した。その結果、SARS-CoV-2 Sは肺の局所、脾臓と小腸の遠位で有意に血管漏出を誘発し、肝臓と脳ではdextran-680の蓄積により、傾向は見られたが有意な漏出ではなかった(図3C-HおよびS2A-D)。

B PBS(n = 27)、DENV2 NS1(15 μg; n = 5)、S(10 μg; n = 25)、およびS(25 μg; n = 5)で処置したマウスからのAを定量化した。
C SARS-CoV-2 S全身血管漏出アッセイからの代表的な肺の画像。マウスにSARS-CoV-2 Sまたはオバルブミン50 µgを指示通りに経鼻投与し、22 hpt後にAと同様にデキストラン680トレーサーを静脈内投与した。デキストラン680投与後2時間(S投与後24時間)マウス臓器を回収し、蛍光スキャナーでデキストラン680の蓄積量を測定した。
D n = 6匹のマウスから得たCの定量化。E 脾臓の代表的な画像を示す以外はCと同じ。F n = 6 マウスからの E の定量化。
G 小腸の代表画像を示した以外はCと同じ。H n = 5 マウスからのGの定量化。
I, J SARS-CoV-2 WA/1分離株の100 TCID50ユニット感染後7日目のK18-hACE2マウスの肺切片にヘマトキシリン・エオジン(H&E)染色を行った。模擬感染させたn=3匹のマウス(I)およびSARS-CoV-2を感染させたn=4匹のマウス(J)の肺の代表画像を表示する。矢印は分散した赤血球を示す。K SARS-CoV-2マウス適応株(MA-10)の指定用量を7日間感染させたC57BL/6マウスの代表的な肺の画像。Aと同様にdextran-680トレーサーを静脈内投与し、dextran-680投与2時間後に肺を採取、ホルマリンで一晩固定し、蛍光スキャナーで蛍光蓄積量を測定した。L 2×104 PFU 条件を除き、n = 4匹のマウスから得た K の定量。MFIは平均蛍光強度。すべてのデータは平均値+/- SEMでプロットされ、両側不対t検定により*p < 0.05, **p < 0.01, ***p < 0.001とした。ソースデータはSource Dataファイルとして提供される。
SARS-CoV-2感染が生体内で血管漏出を引き起こすかどうかを調べるため、SARS-CoV-2のヒト分離株WA/1をK18-hACE2トランスジェニックマウスに感染させた。マウスは病気のピーク時に犠牲にし、マウスの肺と小腸の切片を固定し、ヘマトキシリン・エオジン(H&E)染色を施した。SARS-CoV-2感染マウスの肺と小腸には、対照マウスに比べ有意に炎症細胞が流入していることが観察された。さらに、感染マウスでは、コントロールマウスと比較して、肺や小腸全体に赤血球が分散していることが観察された(図3I、J、S3E、F)。SARS-CoV-2の生体内血管漏出を定量的に調べるために、SARS-CoV-2のマウス適応株(MA10)を感染させたC57BL/6 Jマウスの肺におけるdextran-680の蓄積を測定した。感染後7日目の発病ピーク時に蛍光トレーサーdextran-680を静脈内投与し、マウスの肺における蛍光蓄積量を評価した。その結果、感染マウスでは対照マウスに比べて有意な蛍光シグナルの蓄積が認められ、それはウイルス接種量と正の相関があった(図3K, L)。これらのことから、SARS-CoV-2感染により、マウスのバリア機能障害や血管漏出が誘発されることが明らかとなった。
Sを介したバリア機能障害にはグリコサミノグリカンが必要である
SとHSPGの相互作用がSARS-CoV-2の感染性を高めることから、我々は細胞表面のGAGもSを介したバリア機能障害に必要であると仮定した。ヘパラン硫酸に類似した高電荷の直鎖多糖であるヘパリンがSによる内皮過透過に拮抗する能力を測定したところ、TEERアッセイでヘパリンがSによる内皮過透過を減少させたことから、細胞表面の硫酸化糖鎖との相互作用がSによるバリア機能不全に寄与しているという我々の仮説が裏付けられた(図4A)。Sが介在するバリアー機能障害への糖鎖の寄与をさらに検証するために、内皮細胞表面から特定の糖鎖を除去する組換え酵素を用いた。HS(ヘパリンリアーゼ)、HA(ヒアルロニダーゼ)、CS(コンドロイチナーゼ)、シアル酸(ノイラミニダーゼ)などがあり、これらの条件でSのバリアー機能障害誘発能を検証した。我々は、HS、HA、CSの除去は、シアル酸ではなく、Sによる内皮の過透過性を阻害することを見いだし、TEERで測定したSによるバリア機能障害におけるGAGの役割をさらに支持した(図4BとS4A)。HSとHAの除去もまた、細胞表面のシアル酸レベルで測定されるように、Sを介したEGL破壊を無効にするのに十分であった(Fig.)この表現型に対するHSの寄与を遺伝学的に調べるために、我々はCRISPR-Cas9技術を利用して、HSプロテオグリカン生合成経路に関与する2つの遺伝子、すなわちSLC35B2とXYLT238を個別にノックアウトした細胞株を作った39, 40, 41.を作成した。これらのKO細胞株は、NT対照と比較して、細胞表面に検出可能なHSが少ないことを確認した(図S4B, C)。これらの細胞株をSで処理すると、NTコントロールに比べてSを介したバリア機能障害に対する感受性が低いことがわかり、この経路にGAGが関与していることをさらに証明した(図4E, F)。

B TEER阻害アッセイでは、HPMECの単層をリコンビナントヒアルロニダーゼ(10 μg/mL)、ヘパリンリアーゼIとIII(各5 mU/mL)、ノイラミニダーゼ(1 U/mL)またはコンドロイチナーゼ(25 mU/mL)で処理し、S(10 μg/mL)処理を併用した。TEERの測定は24時間おきに行った。データは、n = 3生物学的複製から得たものである。
C ヒアルロニダーゼ(10 μg/mL)またはヘパリンリアーゼIとIII(各5 mU/mL)で処理したHPMECを、同時にS(10 μg/mL)で処理し24 hptで固定したEGL阻害アッセイ。
D n = 3生物学的複製からのCの定量化。統計は、示された条件とSのみの対照条件との比較である。
E 指定された遺伝子標的化ガイドRNAをコードするレンチウイルスを導入したHPMECのEGL破壊アッセイからの代表画像。細胞は10 µg/mL Sで処理し、シアル酸は24時間後にIFAで可視化した。
F n = 3生物学的複製からのEの定量化。このパネルのコントロールガイドのデータは、Fig. S2Bと同じ実験によるものである。G EおよびFと同じであるが、示されたガイドRNAを使用する。ノンターゲット(NT)データは、2つの細胞株複製からプールされたものである。このパネルのコントロールガイドデータは、Fig. 7Jと同じ実験から得たものである。すべてのパネルにおいて、シアル酸は小麦胚芽アグルチニンで緑色に、核はヘキストで青色に染色し、スケールバーは50μmとした。MFIは平均蛍光強度。点線は正規化した未処理対照条件である。すべてのデータは平均値+/- SEMでプロットされ、*p < 0.05, **p < 0.01, ***p < 0.001, および n.s. p > 0.05 は Tukeyの多重比較検定付きの一元配置分散分析で、(G)は両面不対t検定で分析されたことを除く。パネルA、B、F、Gの統計は未処理のコントロールとの比較、パネルDの統計はSのみのコントロール条件との比較である。ソースデータはSource Dataファイルとして提供される。
細胞外マトリックス調節成分がSARS-CoV-2のS介在性バリア機能障害に寄与している可能性
次に、フラビウイルスNS1を介したEGL破壊に重要であると報告されている成分が、Sを介したEGL破壊に寄与している可能性があると仮定した。そこで、カテプシンL、ヘパラナーゼ(HPSE)、A Disintegrin And Metalloprotease 17(ADAM17)、IL-6R、Matrix Metalloproteinase 9(MMP9)など、ECMの恒常性を制御することが知られているいくつかの内在性酵素や因子が関与していることを検討した。カテプシンL、HPSE、MMP9はフラビウイルスNS1を介した内皮バリアー機能障害に必要である30,31,42,43。MMP9は、ECMを調節するTGF-βの放出に関与していることが報告されている44。ADAM17は、ACE2の遊離、IL-6シグナル、TGF-βシグナルの調節を通じて、COVID-19の病態の調節に関与していることが報告されている45,46。CRISPR/Cas9を用いて、これらの各因子のKO HPMECを作製したところ、HPSE、ADAM17、MMP9を遺伝的に欠損したHPMECは、Sを介したバリア機能障害に対してもはや感受性がないことが判明した。一方、カテプシンLとIL-6Rを欠損したHPMECは、NTコントロールHPMECと同等のバリア機能障害を示した(図4Gおよび図S4D-H)。これらのデータは、Sが介在するバリア機能障害にはEGLの重要なモジュレーターが必要であることを強調し、SとフラビウイルスNS1がバリア機能障害を媒介する違いをさらに明らかにした。
転写解析により、SARS-CoV-2 Sが細胞外マトリックスの恒常性に関与する遺伝子を調節していることが明らかになった。
Sが内皮細胞に及ぼす影響についてさらに理解を深めるため、S処理後のHPMECおよびHPMEC/ACE2のグローバルな転写プロファイルを測定するためにRNAシーケンス(RNA-seq)を実施した。S処理したHPMECでは、未処理のコントロールと比較して、45個のアップレギュレーションと20個のダウンレギュレーションを含む65個の差次的発現遺伝子(DEG)を同定した。S処理したHPMEC/ACE2では、未処理のコントロールと比較して、34個のアップレギュレーションと7個のダウンレギュレーションを持つ42個のDEGが同定された。さらに、HPMECとHPMEC/ACE2から得られたDEGは類似しており、ACE2の発現がSに対するHPMECの転写反応にほとんど影響を与えないことが示された(図5A、B、S5A、B、表S1、S2)。プロテオグリカン、コラーゲン、インテグリンなどのECM構成要素をコードする遺伝子や、内皮性システインプロテアーゼであるCAPN2(カルパイン)、フィブロネクチンとプロテオグリカンの分解を担うHTRA1などのECM分解に関与するタンパク質の遺伝子も多数認められ、SがEGL47を破壊しているという我々の観察結果と一致している。CS、HS、デルマタン硫酸プロテオグリカンの生合成を担う酵素をコードするXYLT2 (xylosyltransferase 2) の発現増加は、Sによるバリア機能不全後のEGL回復経路に関与する可能性がある。トランスクリプトームプロファイリングは、TGF-βシグナル伝達経路の活性化も示し、TGFBI、LTBP2、LTBP3、SERPINE1、FBLN5、POSTN、FN1、THBS1、BGN、ITGB5、ITGA4遺伝子の発現上昇によって実証された。TGF-βは、細胞の分化、増殖、移動のメディエーターとしてよく知られており、血管透過性の調節や炎症反応に重要な役割を担っている48。次に、これらのDEG間の関係を調べるために、Sで処理した親のHPMECから発現上昇した遺伝子のタンパク質-タンパク質相互作用ネットワーク解析を行った。その結果、Sで発現が増加した遺伝子は、タンパク質相互作用が予測される高度なマトリックスを形成しており、ECMまたはECM関連タンパク質の31遺伝子がピンポイントで見つかった。その中には、バリア細胞の細胞骨格とECMをインテグリンを介してつなぐfocal adhesion complexの多数のコンポーネントが含まれていた49 (Fig. 5C)。これらのことから、糖鎖の必要性に加えて、Sが介在するバリア機能障害にはインテグリンが必要であり、インテグリンがTGF-βの成熟とシグナル伝達を促進するという仮説が立てられた。
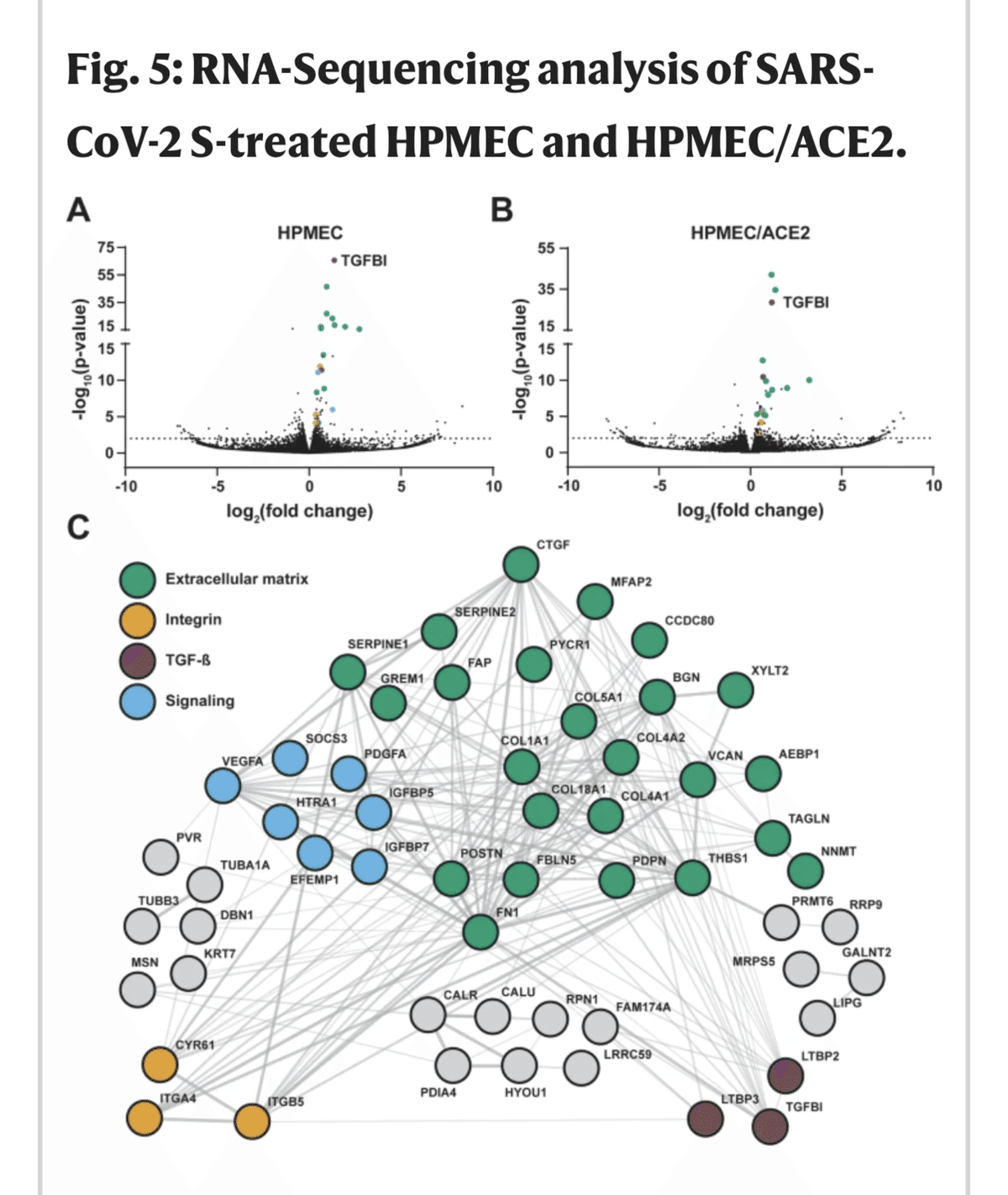
B Aと同じだが、10 µg/mL SARS-CoV-2 Sで処理したHPMEC/ACE2からのDEGを表示。点線は有意性の閾値を示す。DEGの統計的有意性は、Wald検定とBenjamini-Hochberg(BH)p値調整を用いて決定された。
C HPMECのS処理条件と未処理条件の間で同定されたDEGのSTRINGタンパク質-タンパク質相互作用ネットワーク。
Sを介したバリア機能障害にはインテグリンが必要である
インテグリンは、細胞とECMを繋ぎ、バリア機能を維持するために重要な膜貫通タンパク質である50。インテグリンは、ECMの恒常性を制御する因子と相互作用する。例えば、潜在性関連ペプチド(LAP)は、TGF-βと非共有結合し、不活性型に維持される51。LAPは、機械的ストレスなどの様々な刺激に応答して、あるいはインテグリン結合因子との競合を通じて、活性型TGF-βを放出する(その結果、ECMからLAPが放出される)51。インテグリンは、相互作用するタンパク質上の小さなペプチドモチーフ(RGD)を介して、これらの相互作用を媒介する51,52。組換えRGDペプチドは、LAPやTGF-βのような他のRGD含有タンパク質とインテグリン結合を競合できるインテグリン結合モチーフとして一般的に使用されている53。さらに、SARS-CoV-2 Sは、そのRBD19内にRGDモチーフを進化させていることが報告されている。しかしながら、SARS-CoV-2 Sはインテグリンに結合することが示されているが、RGDモチーフの機能的な関連性は不明である18。Sが介在するバリア機能障害におけるインテグリンの役割を調べるために、TEERアッセイとEGL破壊アッセイの両方で、インテグリン阻害剤(ATN-161)がSが介在するバリア機能障害に拮抗する能力を測定した。その結果、ATN-161で処理した細胞は、用量依存的にSを介したバリア機能障害をコントロール条件と比較して抑制することがわかったが、バリア機能には単独では影響を及ぼさなかった(図6A、B、S6A、B)。Sを介したバリア機能障害のインテグリンへの依存性は、HPMECとHPMEC/ACE2細胞の両方でATN-161によってSを介したTEER減少が同等に阻害されたことから、ACE2の発現には依存しなかった(Fig. 6A)。Sを介した内皮過透過性におけるインテグリンの役割をin vivoで確認するために、我々の皮内モデルマウスでATN-161が漏出を抑制する能力をテストした。その結果、ATN-161はPBSコントロールと比較して、Sによる血管漏出を有意に抑制したが、それ自身は漏出を誘発しなかった(図6C, D)。次に、SARS-CoV-2のS RBD内のRGDモチーフを模倣した組み換えRGDペプチドが、HPMECの内皮過透過性とEGL破壊を媒介するのに十分であるかどうかを検証してみた。陰性対照として、KGDペプチド(SARS-CoV-1 Sの対応する配列を模倣)とDRGスクランブルペプチドを使用した。興味深いことに、RGDはSと同様に内皮の過透過とEGLの破壊を引き起こすのに十分であり、一方、KGDとDRGの障壁機能に対する効果は未処理の対照条件と同等であった(図6E、F、およびS6C)。次に、皮内漏出モデルにおいて、RGDペプチドがin vivoで血管漏出を誘発する能力をテストしたところ、対応するin vitroのデータと一致して、RGDペプチドは用量依存的に、完全長Sと同等のレベルで血管漏出を誘発するのに十分であることがわかった (Fig. 6G, H) 。インテグリンの役割を遺伝学的に確認するために、CRISPR-Cas9を用いて、Sと相互作用することが示されている2つのRGD結合インテグリン、integrinα5(α5)およびintegrinβ1(β1)をノックアウトした(Fig.6I)。我々は、Sを介したバリア機能障害は、ITGA5 KO HPMECSではNTコントロールと比較して相対的バリア機能が低いという注意点を除き、NTコントロールのHPMECと比較して両方のKO HPMECラインで著しく阻害されることを見出した(図6J、K、およびS6D)。SARS-CoV-2感染による血管漏出におけるインテグリンの役割をin vivoで調べるために、再びC57BL/6マウスにSARS-CoV-2 MA10を感染させ、感染期間中は毎日インテグリン阻害剤ATN-161を投与した。その後、dextran-680を投与し、感染後7日目のマウスの漏出を評価した。その結果、前回と同様に、マウスへの感染は、模擬感染状態に比べて肺に大きなリークを引き起こすことがわかった。注目すべきは、ATN-161を毎日投与しても、バックグラウンドレベルの血管漏出には影響がなかったが、SARS-CoV-2トリガーによる漏出は有意に抑制されたことで、Sトリガーによる漏出と同様に、ウイルスによる血管漏出はインテグリンを必要とすることが示された(図6L, M)。これらのデータを総合すると、インテグリンはSおよびウイルスが媒介するバリア機能障害に重要な役割を果たし、RGDペプチドは過透過を引き起こすのに十分であることが示された。

B Aと同様にSとATN-161で処理したHPMEC単層の表面上のシアル酸を検出するEGL阻害アッセイ。データは、n = 4(水)、n = 5(ATN-161 0.1 μMと1 μM)、n = 3(ATN-161 10 μM)の生物学的複製からのものである。
C 示した処理を行ったマウスの皮内リークアッセイからの代表的なバック;S(15 µg)とATN-161(1 µM)を同時に注入した。
D n = 7マウスからのCの定量化。E 0.4μMのペプチドで処理したHPMECおよびHPMEC/ACE2単層膜のTEERアッセイ。TEERの測定は、24時間後に行った。データは、n = 3生物学的複製から得たものである。F Eと同じだが、HPMEC単層表面上のシアル酸を検出するEGLアッセイ。データはn = 3 biological replicatesから得た。
G S (15 µg)とRGDペプチドを指定された用量で処理したマウスの皮内漏出アッセイからの代表的なバック。H n = 4匹のマウスから得たGの定量化。
I 指定されたレンチウイルス-エンコーディングガイドRNAで形質転換したHPMECのウェスタンブロット分析。アクチンをローディングコントロールとして使用した。データは、n = 3生物学的複製からの1つの代表的な実験である。
J Iで示した遺伝子を標的とするレンチウイルス-エンコードガイドRNAを導入したHPMECのTEERアッセイ。細胞は10μg/mLのSで処理し、TEERは24時間後に読み取った。データはn = 3生物学的複製から得たものである。
K Jと同様に、形質導入されたHPMECの細胞表面上のシアル酸を検出するEGLアッセイ。
L SARS-CoV-2マウス適応株(MA-10)の2×104PFUで7日間感染させたC57BL/6マウスの代表的な肺の画像である。マウスに10 mg/kgのATN-161、またはビヒクル対照を毎日腹腔内投与した(合計8回)。感染後7日目にdextran-680トレーサーをマウスに静脈内投与した。デキストラン-680投与2時間後に肺を採取し、ホルマリンで一晩固定した後、蛍光スキャナーで蛍光蓄積量を測定した。
M n = 5匹のマウスのLを定量化した。MFIは平均蛍光強度。点線は正規化した無処置対照条件。すべてのデータは、平均値+/- SEMとしてプロットされ、*p < 0.05, **p < 0.01, ***p < 0.001, および n.s. p > 0.05 by One-Way ANOVA with Tukey's Multiple comparisons test except which (D, H, and J) were analyzed by two-sided unpaired t-test。パネルE、F、J、Kの統計は未処理のコントロールとの比較、パネルA、Bの統計はSのみのコントロールとの比較である。
TGF-βシグナルはSARS-CoV-2 Sが媒介するバリア機能障害に必須である
インテグリンは、TGF-βの成熟とシグナル伝達の重要なレギュレーターである48,54。SARS-CoV-2 Sが介在するバリア機能障害におけるインテグリンの重要な役割と、Sが介在するTGF-β転写のアップレギュレーションを明らかにした我々のRNA-Seqデータを考えると、SARS-CoV-2 SのRGDモチーフはインテグリンを巻き込み、成熟TGF-βを放出し、それがTGF-β受容体(TGFBR)と相互作用してシグナルを活性化しバリア機能障害につながるという仮説であった。この仮説を検証するため、Sで処理したHPMECの上清中のTGF-βのレベルを測定したところ、未処理の対照条件と比較して、S処理細胞の上清中のTGF-βが有意に増加していることが観察された(図7A)。このTGF-β産生の機能的帰結を決定するために、HPMECをリコンビナントTGF-βで処理したところ、これは内皮の過透過性を引き起こすのに十分であることがわかった(図7B)。S媒介性の内皮機能障害に対するTGF-βシグナル伝達の寄与を決定するために、TGFBR1の抗体遮断を介してTGF-βシグナル伝達を拮抗させ、抗TGFBR1で処理した細胞はIgGアイソタイプコントロール条件と比較してS媒介性の内皮過透過性に弱いことが分かった(図7Cおよび図S7A)。重要なことは、抗TGFBR1抗体は、それ自身では内皮透過性亢進を引き起こさないことであった(図S7A)。次に、TGF-βシグナル伝達の低分子阻害剤(SB431542)を利用し、この分子で処理した細胞およびマウスは、ビヒクル対照と比較して、S媒介TEER減少、EGL破壊および血管漏出に反応しにくいことがわかった(図7D-G、S7B、C)。最後に、CRISPR-Cas9を利用してTGFBR1 KO HPMECsを作製した(図7H)。これらのKO細胞は、NT対照細胞と比較して、Sを介したバリア機能障害に対する感受性が低いことが分かった(図7I、J、およびS7D)。これらのデータは、SARS-CoV-2 SがHPMECからのTGF-βの放出を促進することを示し、Sによるバリア機能障害におけるTGF-βシグナルの重要な役割を強調するものであった。

B 表示濃度のHPMECのバリア機能に対する組換えTGF-βの効果を測定するTEERアッセイ。TEERの測定値は24時間後に測定された。データはn = 3生物学的複製からのものである。
C 抗TGFBR抗体が、HPMECおよびHPMEC/ACE2のS介在性内皮過透過性(10μg/mLでS)を阻害する能力を測定するTEERアッセイ。TEERの測定は24時間おきに行った。データはn = 3生物学的複製からのものである。
D TGFBR阻害剤SB431542(1μM)がS(10μg/mL)機能を阻害する能力を測定したTEERアッセイ。データはn = 3生物学的複製から得たものである。
E シアル酸を測定するEGLアッセイを除き、Dと同じ。データはn = 3生物学的複製から得たものである。
F 示された処理を行ったマウスの皮内漏出アッセイからの代表的な背部;S(15μg)およびSB431542(1μM)が同時に注入された。
G n = 8匹のマウスから得たFの定量化。
H 標記遺伝子を標的とするガイドRNAをコードするレンチウイルスを導入したHPMECのウェスタンブロット解析。アクチンをローディングコントロールとして使用した。データは、n = 3生物学的複製からの1つの代表的な実験である。
I Hと同じHPMECを10 µg/mLのSで処理し、24時間後に測定したTEERアッセイ。データはn = 3生物学的複製からのものである。J 10μg/mLのSで処理し、24時間後に画像化したHのHPMECのEGL破砕アッセイ。このパネルのコントロールガイドのデータは、図4Gと同じ実験からのものである。データは、n=3生物学的複製物からのものである。
K SARS-CoV-2 Sがバリア機能障害を誘発するACE2非依存性経路をまとめたグラフの要旨。実線は直接的な実験的証拠のある段階を表し、点線は仮説的な段階を表している。すべての図において、グラフの点線は未処理の対照条件を正規化したものである。MFIは平均蛍光強度。すべてのデータは平均値 + /- SEM でプロットされ、*p < 0.05, **p < 0.01, ***p < 0.001, n.s. p > 0.05 は Tukey の多重比較検定付きの一元配置分散分析で、但し (G) は両側 unpaired t-test で分析された。パネルB、D、I、Jの統計は未処理のコントロールとの比較、パネルCとEの統計はSのみのコントロール条件との比較である。ソースデータはSource Dataファイルとして提供される。
考察
我々の研究は、SARS-CoV-2のSがin vitroの上皮細胞および内皮細胞のバリア機能障害とin vivoの血管漏出を媒介する能力とメカニズムを明らかにし、Sだけがウイルス感染とは別にバリア機能障害を媒介できることを示唆している55。我々の研究によると、COVID-19患者の臨床サンプルで観察されたSのレベルは、バリア機能障害を引き起こすのに十分である(2.5 µg/ml)56。この結果から、Sはウイルス侵入に関与するだけでなく、GAGやインテグリンとの相互作用により、TGF-β経路の活性化を介して血管漏出を引き起こすことが示唆された18,57。さらに、我々の研究は、COVID-19の発症時にTGF-βが過剰に産生され、それが病気の重症度と相関していることのメカニズム的説明を提供するものである58,59。このように、我々の研究は、Sが引き金となるバリア機能障害と血管漏出の新しいメカニズムを定義し、COVID-19治療のための新しい治療法の可能性を見いだしたのである。
本研究で使用したSの濃度は2.5〜20 µg/mLであり、大半の実験は10 µg/mL(〜50 nMに相当)で行われた。この濃度は、患者の循環血中に検出されるレベルや、重症のCOVID-19患者の喀痰から検出されるウイルス量(ng/mLからμg/mLのレベル)から推定されるレベルと一致する56,60,61,62,63,64。また、組織深部の毛細血管に蓄積されるSの局所濃度は、患者の血清に循環するレベルよりも高い可能性が高いと仮定している。したがって、我々の研究で利用したSの濃度は、重症のCOVID-19患者に見られる循環レベルと一致する。しかし、SARS-CoV-2感染時に内皮細胞や上皮細胞と相互作用してバリア機能障害を引き起こすSの発生源は、まだ不明である。しかし、SARS-CoV-2感染時に内皮細胞や上皮細胞と相互作用し、バリア機能障害を引き起こすSは、ビリオンに結合した完全長S、可溶性三量体S、そしてリコンビナントRBDであることが示唆された。したがって、SARS-CoV-2は、(1)ウイルス透過性細胞への感染時、(2)細胞上のACE2との相互作用による酵素切断後の可溶性S1の排出、(3)感染細胞表面でのSの発現による隣接細胞との相互作用、(4)ACE2陰性の非透過性細胞との相互作用などの複数の経路でバリア機能異常を引き起こすことが示唆された。これらの可能性を探るためには、in vivoの実験だけでなく、臨床サンプルを用いたさらなる調査が必要である。
Sを介したバリア機能障害に必要な宿主因子を定義する我々の遺伝子データに基づき、SARS-CoV-2のSが、SARS-CoV-220によって特異的に獲得されたRBDの正電荷面を介して細胞表面のGAGに最初に結合するというモデルを提案する。細胞表面に結合したSは、RBD内のRGDインテグリン結合モチーフを介して、α5β1などのインテグリンと結合することができる18。インテグリンと結合すると、定常状態ではTGF-βを不活性状態に維持するLAPを置換し、成熟TGF-βを放出させ、さらにTGFBRに結合して一過性のバリア機能障害を制御するシグナル伝達経路を仲介する(図7K)。このバリア機能の低下は、EGLと細胞間結合複合体の破壊に異なる役割を持つHPSE、ヒアルロニダーゼ、ノイラミニダーゼ、MMP9、ADAM17といった主要酵素の活性化の結果であると思われる。さらに、MMP9とADAM17は、TGF-βの成熟を媒介する別の役割が報告されており、このプロセスを通じて、Sが媒介するバリア機能障害に寄与している可能性もある44,45。この経路は、S細胞結合におけるヘパラン硫酸の役割、Sを介した内皮細胞の活性化とバリア機能不全におけるインテグリンの役割、COVID-19疾患の重症度の相関因子としてのTGF-βを報告している他の研究者の見解によって裏付けられている20,28,58,59。この研究は、SARS-CoV-2 Sがバリア破壊を引き起こすメカニズムに光を当て始めたが、さらなる宿主因子の定義とこの経路に対する各因子の相対的寄与を決定するための研究が必要である。
今回の研究により、ACE2との結合やウイルスの侵入以外にも、Sの新たな役割が明らかになったが、多くの重要な問題が残されている。まず第一に、Sが介在するバリア機能不全がSARS-CoV-2感染の結果にどのように影響するのかということである。以前のデータでは、NS1を介した血管漏出が亜致死性のDENV感染を悪化させることが示されており、可溶性NS1がDENVの病原性を促進することを直接証明している29,65。DENV NS165によって直接引き起こされる炎症反応に加えて、NS1が病原性を促進する一つの仮説は、血液感染したフラビウイルスが血液から高力価で複製できる遠位組織への播種を促進することである32, 42, 55, 66.。したがって、Sを介したバリア機能不全がCOVID-19の病因に寄与する可能性は、SARS-CoV-2の肺から血液、そしてウイルス透過性細胞が存在する遠位臓器への播種を促進することだと推測される。このことは、マウスの肺にSを投与すると、脾臓や小腸で全身漏出が起こるという観察に例示されている(図3)。このことは、COVID-19患者に見られる多様な臨床症状を説明するのに役立ち、実際、急性COVID-19後遺症を持つ患者においてSの持続的な循環が報告されている61。さらに、肺の表面は多くのプロテオグリカンや糖タンパク質からなる高密度のグリコカリックスで覆われており、その主成分は膜結合型およびゲル形成型のムチンからなる粘液である。最近、これらのムチンは、ウイルスと細胞の相互作用を立体的に阻害することにより、細胞がSARS-CoV-2感染に対して不応性となることを証明した。したがって、EGLを破壊するSによる障壁機能障害は、ウイルス透過性の上皮細胞を、侵入するウイルスに対してよりアクセスしやすくするのかもしれない67,68。
SARS-CoV-2のSはHPMECの内皮過透過性を引き起こすが、HCoV-229EやHCoV-OC43のSは引き起こさないという我々の観察は、これらの肺細胞のバリア機能障害を引き起こす能力は、すべてのコロナウイルスに等しく保存されていないことを示唆するものであった。我々は、SARS-CoV-2 SがHPMEC上のヘパラン硫酸やインテグリンと相互作用する能力が高いことがこの特異性を説明しているのではないかと考えているが、この可能性を検証するためにはさらなる研究が必要である18,20。さらに、細胞表面でのACE2の発現は、Sタンパク質が内皮細胞や上皮細胞と相互作用し、バリア機能障害を引き起こす能力に影響を与える可能性がある。これは、SARS-CoV-1やHCoV-NL63が、ACE2を侵入受容体として利用していることに起因しているのかもしれない69,70。SARS-CoV-1 と HCoV-NL63 の両方の S が Vero-E6 細胞と相互作用すると、別のメカニズムではあるが、ACE2 の発現が低下することが示されており、これが SARS-CoV-1 S21,23,71 の場合の組織傷害に寄与していることが示されている。重要なことは、SARS-CoV-2 Sもまた、ACE2依存的に炎症反応を引き起こし、バリア機能を障害することが、いくつかの報告で明らかにされている24,25,27,72,73.ACE2非依存的経路とACE2依存的経路のin vivoでの血管漏出への相対的寄与を理解し、あるコロナウイルスのSタンパク質がどの経路を引き起こすかを明確にすることが重要であると思われる。
Sが内皮機能障害と血管漏出を引き起こすのに十分であるという我々の観察により、フラビウイルスのNS1タンパク質と直接比較することが可能になった。このような比較は、ウイルスがどのようにシグナル伝達経路を活性化してバリア機能障害を媒介するのかについての理解に役立ち、複数の可溶性ウイルスタンパク質を標的とした汎用の抗漏出治療薬の開発という概念につながるものである。SとNS1が内皮バリア機能障害を引き起こすメカニズムには、類似点と相違点の両方があることが明らかである55。我々のメカニズム調査では、GAG(HS、CS、HA)、MMP9、ADAM17、HPSE、インテグリン、TGF-βシグナルがSが媒介するバリア機能障害に寄与していることが明らかにされた。GAGの結合とMMP9、HPSE、ヒアルロニダーゼ、ノイラミニダーゼなどの酵素の活性化は、NS1-とSを介した内皮機能障害の両方に共通する必要条件である。一方、カテプシンLはNS1の発症にのみ必須であり30,31、Sを介した機能障害にはインテグリンの関与とTGF-βシグナルが必要であるようである。これらの相違は、NS1がバリア機能障害のピークをNS1投与後6時間までに引き起こすのに対し、Sが介在する場合は24時間までにピークを迎え、6時間後にはバリア機能の障害は観察されないという、我々のin vitro過透水アッセイの異なる動態を説明することができるかもしれない。この理由の一つは、Sを介した内皮機能障害にはセカンドメッセンジャーとしてTGF-βの産生とシグナル伝達が必要であるが、フラビウイルスNS1による漏出には必要ないためと思われる。これらの経路が類似していながら異なるのはなぜかを完全に理解するためには、SとNS1を介したバリア機能不全のメカニズムのさらなる比較研究が必要である。
インテグリンとTGF-βはSを介した血管漏出に必要であるが、in vivoでのSARS-CoV-2感染の調節におけるそれらの役割は複雑であることは間違いない。例えば、免疫細胞の接着/浸潤、炎症反応の調節、組織修復におけるインテグリンとTGF-βのよく知られた役割は、ヒトにおけるSARS-CoV-2ウイルス感染に複雑な影響を及ぼすと思われる74,75。さらに、COVID-19における血管漏出の病原性に加えて、肺のバリア機能障害は、ウイルスを除去できる免疫細胞の浸潤を促進し、宿主にとって有益であると予測される。しかし、これらの免疫細胞の過剰活性化は、重症COVID-19のARDSと通常関連する「サイトカインの嵐」を引き起こす可能性がある。SARS-CoV-2のin vivo感染における血管漏れの影響の違いを明らかにするためには、さらなる研究が必要である。また、報告されているCOVID-19の病状は多様であり、免疫細胞の浸潤やウイルス感染による肺細胞障害など、血管漏出以外の要因で説明できる可能性があることを考慮することが重要である。COVID-19の重症度に対する血管漏れの相対的な寄与を理解することは、間違いなく複雑であるが、それでも重要な問題である。
我々の作業仮説は、Sが単独で病態を媒介するのではなく、Sによって引き起こされる可逆的な血管漏出が、感染患者の遠位組織へのSARS-CoV-2のウイルス播種を促進し、それが重度の疾患発現につながる可能性があるということに注意することが重要である。しかし、Sを単独で投与したマウスでは、著しい血管漏出が観察されたものの、病的な兆候をあからさまに示すことはなかった。重要なことは、我々の表現型が重症のCOVID-19症例で観察されるレベルに類似したng-μg/mLレベルを必要とすることから、COVIDワクチン接種後の患者の循環するSの量(pg/mLレベル)は、血管漏出を引き起こすには低すぎることが示唆された56,60。以上のことから,我々の研究および入手可能な文献76は,Sを介した血管漏れはCOVID-19ワクチン接種によって生じることはなく,したがって,いかなるワクチン有害事象とも関連しないことを示している.
以上のように、本研究はCOVID-19に関連した血管漏出におけるSの役割を明らかにし、ウイルス感染やACE2受容体とは独立してSがこのプロセスを仲介する仕組みについて洞察を与えている。このメカニズムの構造的基盤や、この経路がSARS-CoV-2感染やヒトの疾病に与える影響についてはまだ多くの研究が必要であるが、この研究はCOVID-19関連血管漏出に対するSの寄与を明らかにし始め、将来の研究のための基礎を提供するものである。
研究方法
マウス
6-8週齢の野生型C57BL/6 JおよびK18-hACE2 [B6.Cg-Tg(K18-ACE2)2Prlmn/J] マウスの両性はJackson Laboratoryから購入し、カリフォルニア大学バークレー校動物施設にて特定の病原体を含まない条件下で飼育された。マウスは、12時間の明暗サイクルで温度制御された環境下に置かれ、餌と水は自由に与えられる。すべての実験および手順は、UC Berkeley Animal Care and Use Committee、プロトコルAUP-2014-08-6638-2およびAUP-2020-07-13458によって事前承認され、連邦および大学の規制を遵守して実施された。
細胞株
レンチウイルス産生に使用したHEK293T細胞はATCCから入手し、10%FBS(コーニング)及び1%ペニシリン/ストレプトマイシン(ギブコ)を補充したDMEM、高グルコース及びGlutaMAXTM(ギブコ)(D10培地)において37℃、5%CO2で維持した。Calu-3ヒト肺上皮細胞は、UC Berkeley Cell Culture Facilityから入手し、D10培地中、37℃、5%CO2で維持した。ヒト肺微小血管内皮細胞(HPMEC)[ラインHpMEC-ST1.6 R]は、ドイツのヨハネス・グーテンベルク大学のJ.C. Kirkpatrick博士から贈られたものである。これらの細胞は成人男性ドナーから単離され、肺内皮細胞のすべての主要な表現型メーカーを示す不死化クローンが選択された77。HPMECは、Endothelial Cell Growth Medium-2 (EGM-2TM) supplemental bullet kit (Lonza)で補充したEndothelial Cell Growth basal medium 2で培養された。細胞は37℃、5%CO2で維持した。本研究では、ヒトACE2(hACE2)遺伝子を過剰発現するHPMEC(HPMEC/ACE2)、およびいくつかのノックアウト細胞株を含む、これらの親細胞のいくつかの遺伝子改変バージョンを作製した。hACE2をコードするプラスミドはHyeryun Choeから贈られた(Addgene plasmid #1786; http://n2t.net/addgene:1786; RRID:Addgene_1786)70.Vero-E6 細胞は SARS-CoV-2 の滴定に使用し、D10 培地を用いて 37℃、5%CO2 で維持した。
リコンビナントタンパク質
全長、安定化SARS-CoV-2 S ectodomainおよびRBD(Wuhan-Hu-1の配列に基づく)78をコードする配列は、以前に報告したように、安定に形質転換した293細胞から発現、精製した79。精製タンパク質は、PBSに1 mg/mLで調合し、アリコートで-80℃に保存した。リコンビナントDENV血清型2 NS1は、Native Antigen Companyから購入し、以前に特性評価した(Dengue virus serotype 2 NS1 [accession # P29990.1, Thailand/16681/84])36。SARS-CoV-2 S安定化三量体は、Native Antigen Companyから、B.1.1.7 (Alpha variant, product #REC31924), B.1.351 (ベータ・バリアント、製品番号REC31963), B.1.1.28/P.1 (ガンマ・バリアント、製品番号REC31944), B.1.617.2+AY.1+AY.2+AY.3 (デルタ・バリアント、製品番号REC31975) および B.1.1.529 (オミクロン・バリアント、製品番号REC32008) を含む、様々なウイルス・バリアントからの三量体を購入しました。リコンビナント VEGF (V7259) および TGF-β1 (100-21) はそれぞれ Sigma および PeproTech から購入し、製造者の指示に従って再懸濁/保存した。
水疱性口内炎ウイルス(VSV)スパイク偽型ウイルスの作製と感染
SARS-CoV-2 S疑似型ウイルスは、G糖タンパク質をレニラ・ルシフェラーゼ・レポーター遺伝子に交換したVSV Indiana血清型完全長相補DNAクローンに由来するVSV-ΔG-rLucシステムを用いて作製した。簡単に言うと、HEK293T細胞の15cm2ディッシュに、45μgの全DNAを用いてSARS-CoV-2 S (Wuhan-Hu-1, Accession #QHD43416.1) をコードするプラスミドをトランスフェクションした。トランスフェクション後24時間で、VSV-ΔG-rLuc疑似型を発現するVSV-Gを使用して、トランスフェクトされたHEK293T細胞に感染させた。トランスフェクション後48時間で培地を採取し、NTEバッファ(150 mM NaCl、40 mM Tris-HCl、1 mM EDTA、pH 8.0)中の20%ショ糖クッションで110,000×g、1.5時間超遠心した。ウイルスの再懸濁のために、NTE Bufferに5%のスクロースを加え、ウイルスのアリコートを-80℃で保存した。
SARS-CoV-2ストックの作製と感染
SARS-CoV-2のストックは、以前に記載されたように製造された80。簡単に言えば、SARS-CoV-2のUSA-WA1/2020株またはマウス適応MA10株をそれぞれBEI Resourcesまたはノースカロライナ大学チャペルヒル校のRalph S. Baric博士81から入手し、0.45μMシリンジフィルターを通過させた。これらのろ過されたストック5μLをVero-E6細胞のT-175フラスコに加え、ウイルス継代1を作製した。細胞障害効果(CPE)を毎日モニターし、〜70%の細胞障害効果が明らかになったとき(〜48 hpi)、フラスコを凍結した。解凍したライセートを集め、細胞残屑を3000rpmで20分間ペレット化した。その後、清澄化したウイルス上清を分注し、感染性ウイルスをTCID50で定量化した。SARS-CoV-2ワーキングストックを作製するために、上記のように、Vero-E6細胞のT175フラスコに継代1のストックを5μLずつ接種した。得られたウイルス力価は、1×106-5×106 TCID50 units/mLであった。マウス感染では、8〜10週齢のK18-hACE2またはC57BL/6雄マウスをイソフルランで麻酔し、図に示した株と用量のSARS-CoV-2を経鼻的に接種した。
レンチウイルスの作製と導入
ヒトACE2または図に示した遺伝子を標的とするCRISPRガイドRNAをコードするレンチウイルス粒子は、以前に報告された第2世代のレンチウイルスシステムを使用して製造した82。簡単に言えば、レンチウイルスベクターを、パッケージングベクター(psPAX2)およびシュードタイピングベクター(pMD2.G)とともに、製造者の指示書に従ってリポフェクタミン3000トランスフェクションプロトコルを用いて293 T細胞にトランスフェクトした。トランスフェクション後12時間後に培地を交換した。トランスフェクト細胞の培地中に放出されたレンチウイルスをトランスフェクション後24時間、36時間、48時間に回収し、プールしたレンチウイルス含有培地を0.45μMシリンジフィルター(ミリポア)で濾過した。標的HPMECをレンチウイルスとともに48時間インキュベートした後、2 µg/mLピューロマイシン(シグマ社)で3回継代選択した。
経上皮/内皮電気抵抗アッセイ
上皮および内皮のバリア破壊(透過性亢進)は、以前に記載したように、経上皮/内皮電気抵抗(TEER)アッセイによって測定した36。簡単に言えば、6×104個のHPMECまたは2×105個のCalu-3を、24ウェルのトランスウェルポリカーボネート膜インサートの頂部チャンバー(Transwell permeable support, 0.4 μM, 6.5 mm insert; Corning)に300μLで播き、1.5mLの培地を基底部チャンバーに添加した。培地は、細胞が最大バリア抵抗で測定される完全な単層を形成するまで(HPMECでは〜3日、Calu-3では〜15日)、頂側および底側チャンバーの両方から毎日交換した。実験当日、各トランスウェルのTEERを測定し、細胞の抵抗レベルが最小値でほぼ同等(5オーム以内)であることを確認した。抵抗値が異常値を示すトランスウェルは、実験から除外した。トランスウェルの頂部チャンバーに処理物(図に示す)を添加した。電気抵抗値は、「箸」電極(World Precision Instruments社製)を備えたEpithelial Volt Ohm Meter(EVOM)を用いて、図に示した時点でオーム(Ω)単位で測定された。未処理細胞のあるインサート、および培地のみを含む細胞のないインサート(ブランク)は、ベースライン電気抵抗を計算するための陰性対照として使用された。相対的TEERは、抵抗値の比率として、(Ω実験条件-Ωブランク)/(Ω未処理細胞-Ωブランク)として算出した。
内皮/上皮糖衣層(EGL)破壊アッセイ
EGL破壊を媒介するタンパク質処理の能力を測定するために、6 × 104 HPMECまたは2 × 105 Calu-3細胞を24ウェルプレートの0.2%ゼラチン(Sigma)コーティングしたガラスカバースリップ上に播種した。3日間(HPMEC)または15日間(Calu-3)、培地を1日おきに交換し、完全にコンフルエントな単層を形成させた。実験当日、図に示す処理物を直接ウェルに添加した。処理物と細胞を指示された時間(一般にS処理では24時間)インキュベートした後、細胞を1x PBSで2回洗浄し、4%ホルムアルデヒド/PBS(Thermo Fisher Scientific)で固定した。カバースリップを、ProLong Gold(Thermo Fisher Scientific)を滴下した顕微鏡スライドに取り付け、Zeiss LSM 710倒立共焦点顕微鏡(CRL Molecular Imaging Center、UC Berkeley)を用いて画像化した。EGL破壊は、Alexa Fluor 647 (Thermo Fisher Scientific, W32466)、ヒアルロン酸 (Abcam ab53842) 、ヘパラン硫酸 (amsbio, clone F58-10E6, 370255-s) またはコンドロイチン硫酸 (Thermo Fisher Scientific, clone CS-56, ma1-83055) に結合したシアル酸特異的レクチン、コムギジャコウネート (WGA) を用いて、細胞上のシアル酸レベルモニターにより評価された。EGL破壊酵素の発現は、ヒアルロニダーゼ(Abcam, clone PH20, ab196596)とノイラミニダーゼ2(Thermo Fisher Scientific, pa5-35114)についてサポニン透過性細胞で評価された。染色は、生細胞1時間前の固定化培地で希釈したWGA-647を100μg/mLで生細胞染色を行った。他の全てのGAG(HA、HS、CS)は、固定した細胞上で、抗体を含む染色バッファーを15μl滴下した上にカバースリップを向け染色した。核はHoechst 33342 (Immunochemistry) を1:200の希釈率で添加することにより染色した。すべての顕微鏡画像は20倍の倍率で撮影された。
インビボ真皮血管漏出アッセイ
S糖タンパク質によって引き起こされる血管漏出を調べるために、以前に記述したように、マウス真皮漏出モデルを利用した36。簡単に言えば、6-7週齢のWT C57BL/6 J雌マウス(Jackson Labsから購入)の背部真皮を剃毛した。3〜4日後、標記処理物(典型的には10μgのSARS-CoV-2 S)を、剃毛したマウス真皮の離散的なスポットに皮内(ID)注射した(50μL/注射部位)。ID注射の直後に、Alexa Fluor 680 (Sigma)と結合した10-kDaデキストランを150μL(合計25μg)静脈内投与した。注射後2時間後にマウスを安楽死させ、背部真皮を切除してシャーレに入れた。蛍光スキャナー(LI-COR Odyssey CLx Imaging system)を用いて、波長700 nmでマウスの真皮における蛍光シグナルの蓄積を可視化した。注入部位周辺の漏出はImage Studioソフトウェア(LI-COR Biosciences社製)を用いて定量化した。低分子阻害剤実験では、ID注入の30分前に阻害剤をSARS-CoV-2 Sと混合した。
In vivo全身性血管漏出
SARS-CoV-2 SまたはSARS-CoV-2感染によって引き起こされる全身性血管漏出を調べるために、我々は以前に記述したように修正した全身性血管漏出アッセイを実施した32。簡単に言えば、50μgのSARS-CoV-2 S、OVAまたはSARS-CoV-2ウイルスストック(図に示す)を6-7週齢のWT C57BL/6 J雌マウス(Jackson Labsから購入)に鼻腔内投与した。指示された感染後22時間ptおよび7日目に、マウスは、Alexa Fluor 680に結合した10kDaデキストラン(150μL、170μg/mL;Sigma)の静脈内注射を受けた。このトレーサー色素を2時間マウスに循環させた後、マウスを安楽死させ、臓器(肺、脾臓、肝臓、小腸、脳)を採取してシャーレに載せた。SARS-CoV-2感染では、臓器は10mLの10%中性緩衝ホルマリン溶液(Sigma)で一晩固定してから撮影した。蛍光スキャナー(LI-COR Odyssey CLx Imaging system)を用いて、波長700 nmで臓器内の蛍光信号の蓄積を可視化した。Image Studio Lite ソフトウェアバージョン5.2(LI-COR Biosciences)を用いてリークを定量化した。
ヘマトキシリン・エオジン(H&E)染色
組織学および H&E 染色は、HistoWiz Inc. (histowiz.com) が標準プロトコルと自動ワークフローに従って実施した。臓器はUC Berkeleyで一晩10%中性緩衝ホルマリン溶液で固定され、その後HistoWizに送られ、そこで処理されてパラフィンに埋め込まれ、4μmの切片が調製された。H&E染色後、切片を脱水し、TissueTek-Prisma and Coverslipper(Sakura)を用いてフィルムカバースリッピングを行った。Aperio AT2 顕微鏡(Leica Biosystems)でスライド全体の走査(40×)を行った.
EGL酵素の調製と消化
Sを介した内皮機能障害に対する糖鎖成分の寄与を調べるために、ヒアルロン酸、ヘパラン硫酸、シアル酸、コンドロイチン硫酸を含む特定の糖鎖成分を消化するために組換え酵素を使用した。リコンビナントヘパリンリアーゼIとIIIはIBEXから入手し、HPMECはそれぞれ5 mU/mLで、リコンビナントヒアルロニダーゼ(Sigma, H3506)は10 μg/mLで、リコンビナントノイラミナーゼ(Sigma, N2876)は1 U/mL で、リコンビナントコンドロイチナーゼABC(Sigma, C3667)は25 mU/mL で処理された。すべての酵素をSと同時にHPMECに添加し、TEER/EGLアッセイを上記のように24hptで実施した。
CRISPR-Cas9ノックアウト
遺伝子特異的ノックアウト細胞株を作製するために、以前に記載したように、Feng Zhangから入手したlentiCRISPR v2 lentivirus construct (Addgene plasmid # 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961) に基づくCRISPR-Cas9パイプラインを活用した83。簡単に言えば、ガイドRNA標的化配列をBrunello CRISPR KOガイドライブラリー84から選択し、lentiCRISPR v2プラスミドにクローニングした。本研究で利用したガイドRNA配列は、表S3に要約されている。レンチウイルスを上記のように製造し、HPMECを2μg/mLのピューロマイシンを含むEGM-2培地で形質導入し、選択で3回継代した。細胞のポリクローナル集団は、タンパク質発現を測定するためのウェスタンブロット、または機能を確認するためのIFAによるHS染色(HS生合成経路遺伝子について)のいずれかによって機能的ノックアウトについて特徴づけられた。
RNAシークエンス
SARS-CoV-2 Sで処理した細胞の転写反応を特徴づけるために、RNA-Sequencingを実施した。簡単に説明すると、HPMECとHPMEC/ACE2を10 µg/mL SARS-CoV-2 Sで処理し、処理後24時間でTRI試薬(Sigma)中で細胞溶解液を回収した。製造者の指示に従い、Direct-zol RNA miniprep kit(Zymo Research)を用いてトータルRNAを抽出した。RNAはQubit Flex Fluorometer(ThermoFisher)を用いて定量し、Bioanalyzer(RNA Pico;Agilent)で品質を測定した。1 µg の RNA を SMARTer Stranded Total RNA Sample Prep Kit - HI Mammalian (Takara Bio) を用いて、メーカーの説明書に従ってライブラリ調製に使用した。調製したライブラリーの品質はBioanalyzer (High Sensitivity DNA; Agilent) で評価し、UCSF Center for Advanced TechnologyでS4フローセルと150塩基対ペアエンドシーケンスを用いてNovaSeq 6000 (Illumina) で配列決定した。サンプルライブラリーのシーケンス後、FastQC (version 0.11.8)85 および MultiQC (version 1.8)86 を用いて、シーケンスリードがリード数および品質に関する事前に設定したカットオフを満たしていることを確認するため、fastqファイルの品質管理を行った。品質フィルタリングとアダプターのトリミングは、BBduk tools (version 38.76, https://sourceforge.net/projects/bbmap) を用いて実施しました。残りのリードは、STAR (version 2.7.0 f)87を用いてENSEMBL GRCh38 human reference genome assembly (release 33)にアライメントし、Subread package88内のfeatureCounts (version 2.0.0)を用いて遺伝子頻度をカウントした。DEGの比較解析は、R(バージョン4.0.3)に実装されたDESeq2(バージョン1.28.1)89で用いられる負の二項分布モデルを用いて行った。Benjamini-Hochberg (BH) 調整のP値閾値0.05を通過したすべての遺伝子が含まれた。DEGの階層的クラスタリングと可視化は、ComplexHeatmap (version 2.4.2) とpheatmapパッケージ (version 1.0.12) を用いて実施した。クラスタリング方法はcomplete linkageを用い、クラスタはEuclidean distanceに基づく。同定されたDEGは、STRINGにより、予測されるタンパク質-タンパク質相互作用ネットワークと、エンリッチドパスウェイ解析(https://string-db.org/)により解析された。
TGF-β 酵素連動免疫吸着アッセイ(ELISA)
S処理した細胞の上清中のTGF-β1のレベルを測定するために、ヒトTGF-β1 DuoSet ELISA(DY240, R&D Systems)を使用した。簡単に言えば、免疫反応性TGF-β1を検出するために、SARS-CoV-2 Sで処理した24時間後に細胞上清を集め、1N HClで活性化した。pH中和後、サンプルとリコンビナントTGF-β1標準をマウス抗ヒトTGF-β1捕捉抗体でコートしたELISAプレートに移し、室温で2時間インキュベートした。その後、プレートをビオチン化ニワトリ抗ヒトTGF-β1検出抗体とインキュベートし、ストレプトアビジン-西洋ワサビペルオキシダーゼ(HRP)、テトラメチルベンジジン(TMB)基質でシグナル検出を行った。450nmに設定したマイクロプレートリーダーを用いて、各ウェルの光学密度を測定した。TGF-β1レベルは、4パラメータ・ロジスティック(4-PL)標準曲線からの内挿により決定した。
多角度光散乱を用いたサイズ排除クロマトグラフィー(SEC-MALS)
可溶性コロナウイルスSとRBDの純度とオリゴマー状態を特徴付けるために、精製タンパク質をSRT SEC-1000 カラム (4.6 × 300 mm, Sepax) にPBS中0.35 mL/分で注入(S用)またはSuperdex 200 Increase カラム (3.2 × 300 mm, Cytiva) にPBS中 0.15 mL/分で注入(RBD用)しました。カラムおよび1260 Infinity II HPLC (Agilent), miniDAWN TREOS II MALS検出器 (Wyatt), Optilab T-rEX 屈折率検出器 (Wyatt) を含むSEC-MALSシステム全体は、分析前に少なくとも24時間PBSで平衡化された。SとRBDの分子量はAstra (Wyatt) を用いて決定した。
低分子阻害剤、ペプチド、抗体
TEERおよびEGL阻害アッセイには、以下の低分子阻害剤および抗体を使用した。ATN-161 (Sigma, SML2079), SB 431542 hydrate (Sigma, S4317), ヘパリン (Sigma, H3393), 抗スパイク (Genetex, 1A9, GTX632604), 抗スパイク (Absolute Antibody, CR3022), ウサギ抗TGFBR1 (Thermo Fisher Scientific, PA5-32631).すべての化学物質と抗体は、製造者の指示に従って再懸濁され、利用された。RGD、KGD、DRGペプチドはシグマ社により合成された。
SDS-PAGE とウェスタンブロット
組換えタンパク質または細胞ライセートをタンパク質サンプルバッファー (0.1 M Tris pH 6.8, 4% SDS, 4 mM EDTA, 286 mM 2-mercaptoethanol, 3.2 M glycerol, 0.05% bromophenol blue) で集め、SDS-PAGEで分離した。次に、タンパク質をニトロセルロース膜に移し、5%スキムミルクを含む0.1% Tween20入りPBS (PBST) で希釈した一次抗体でプロービングした。膜と抗体は4℃で一晩ロッキングしながらインキュベートした。翌日、膜をPBSTで3回洗浄した後、PBST中の5%牛乳で1:5,000に希釈したHRP標識二次抗体で室温で1時間プロービングした。その後、膜をPBSTでさらに3回洗浄してから、自家製ECL試薬で現像し、Image Labソフトウェアバージョン6.01(Bio-Rad)付きのChemiDocシステムで画像化した。本研究では、以下の抗体を使用した。ヤギ抗ACE2(R&D Systems、AF933)、ウサギ抗インテグリンα5(Abcam、ab150361)、ウサギ抗ITGB1(Thermo Fisher Scientific、PA5-29606)、ウサギ抗ヘパラナーゼ1(Abcam、EPR22365-230、ab254254)、ウサギ抗MMP-9(Cell Signaling Technology、#3852)、マウス抗TACE/ADAM17(Santa Cruz Biotechnologies、B-6、 sc-390859),マウス抗カテプシンL(Thermo Fisher Scientific, 33-2, BMS1032)、ウサギ抗TGFBR1(Thermo Fisher Scientific, PA5-32631)、マウス抗His(MA1-21315, Thermo Scientific)。マウス抗β-アクチンHRP(Santa Cruz Biotechnologies、sc-47778 HRP)、ヤギ抗マウスHRP(Biolegend、405306)、ロバ抗ラビットHRP(Biolegend、406401)、ドンキ抗ヒトHRP(Biolegend、410902)。すべての一次抗体は1:1000の希釈で使用し、すべての二次抗体は1:5000の濃度で使用した。
統計
すべてのデータは、GraphPad Prism 8 ソフトウェアを使用してプロットし、定量分析を行った。実験は、他に指示された場合を除き、少なくとも3回繰り返した。実験はポジティブコントロールとネガティブコントロールの両方を用いて計画・実施し、包接・除外判定に使用した。研究者は実験中に盲検化されていない。免疫蛍光顕微鏡の実験では、ランダムフィールドの画像を撮影した。定量分析を伴うすべての実験では、データは平均値±SEMとして表示されている。本研究で使用した統計的検定には、図に示すように、多重比較検定を伴うANOVA分析、およびt検定が含まれる。上記の統計テストの結果のp値は、n.s.(有意ではないp>0.05)、*p < 0.05; **p < 0.01; ***p < 0.001として表示されている。表示されていない統計値はすべて有意でない。
報告書の概要
研究デザインの詳細については、本記事にリンクされているNature Portfolio Reporting Summaryをご覧ください。
データの入手方法
図に関連するすべての生データは、この投稿に含まれているか、またはリクエストに応じて入手可能である。RNA-Seq生データは、BioProject accession # PRJNA807823 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA807823) の一部としてNCBI SRAにアップロードされています。ソースデータは本論文で提供されている。
以下reference省略