生成AIを用いたAI創薬の実践 Part2 - BioNeMoを用いたタンパク質言語モデルの事前学習
この記事は、"生成AIを用いたAI創薬の実践 Part1 -タンパク質言語モデル基礎編"の続編です。
生成AIを用いた創薬の可能性についてPart1で解説をしてきましたが、実際に一から実装しようとすると、マルチノードGPUを用いた分散処理や、モデル構造を理解した効率的な計算が求められるなど、高いエンジニアリング力が必要となってきます。このような背景から、創薬領域における基盤モデルを簡単に活用できるフレームワークNVIDIAのBioNeMoが開発・公開されました。
この記事では、BioNeMoを初めて使用する方を対象に、タンパク質言語モデルの事前学習からファインチューニングまでの実装方法を紹介します。
BioNeMoとは
創薬AIは、創薬のドメインとAIの知識、そして実装力が求められるため、難易度の高い領域です。一方、実装については比較的簡単に行うことができるよう、便利なフレームワークが提供されています。NVIDIAが提供するBioNeMoもそのうちの一つです。BioNeMoは、生物学的な研究をサポートするために開発されたAIフレームワークで、タンパク質や分子の設計に特化しています。BioNeMoは、NeMoフレームワークを基にしており、大規模なトランスフォーマーモデルの開発やトレーニングを簡便に行える機能を提供します。特に、マルチGPUやマルチノードを活用した高度なトレーニングをいくつかの設定を変更するだけで実行することができ、分散学習の大変な部分を抽象化してくれています。

BioNeMoとWandBとのintegration
BioNeMoは、WandBとのインテグレーションを提供しており、大規模な学習からファインチューニング、最適化までWandBで実験管理をしながら開発を進めることができます。BioNeMoはPytorch lightningをもとに開発をされており、BioNeMoにおけるWandBとのインテグレーションはPytorch lightningとWandBのインテグレーション機能(WandbLoggerによるlossのトラッキング)をもとに開発されています。

一方、WandBはモデル学習過程のトラッキング機能だけではなく、Artifactsによるデータやモデルのバージョン管理なども提供しており、Part1のblogで触れたTokyo1でのハンズオンにあたり、独自にBioNeMoとのWandBのインテグレーションを強化したrepositoryおよびドキュメント"Pretraining & Finetuning of Protein Language Model with BioNeMO & WandB JP"を開発・公開しました(公式のBioNeMoコンテナへの統合を現在進めています)。以下では、その強化されたWandBとのインテグレーションも使用したBioNeMoによる事前学習の実装例を解説します。
以下に、実装の流れを示します。
0. まずは環境構築を行います
1. 次にデータを準備し、WandB Artifactsでデータ管理を行います
2. その後、WandB Experimentsで実験管理をしながら、事前学習を行います。
3. 最後に、事前学習したモデルをWandB Registryに登録をします。
今回はファインチューニングの実装例は長くなるため紹介しませんが、サンプルスクリプトには含めているので、ご関心がある方はご確認ください。

タンパク質言語モデルの事前学習の実装
0. 環境構築
BioNeMoの細かい環境設定については、BioNemo Frameの公式ドキュメントを参考にしてください。以下に、この記事で解説されているサンプルコードをを実装するための環境構築手順を示します。
Prerequisites
x86 Linux systems
Docker (with GPU support, docker engine >= 19.03)
Python 3.10 or above
Pytorch 1.13.1 or above
NeMo pinned to version 1.20 NVIDIA GPU, if you intend to do model training.
Tested GPUs: H100, A100, V100 RTX A6000, A8000 Tesla T4 GeForce RTX 2080 Ti (GPUs with known issues: Tesla K80)
bfloat16 precision requires an Ampere generation GPU or higher.
アカウント設定
Weights & Biases Account
Weights & Biasesのアカウントを持っていない方は、Weights & Biases のホームページよりトライアルアカウントを作成してください.
Access to BioNeMo Framework from NVIDIA. ("Get Started With NVIDIA BioNeMo"の"Training Framework"にアクセスできるようにする)
Access to NGC ( NVIDIA GPU CLOUD)
Sign into NGC.
Download and install NGC CLI and setup your API Key
※ NGCはBioNeMoのモデルにアクセスするために必要となります
(Optional) Access to NVIDIA DGX compute infrastructure (DGX-Cloud or DGX-Pod). (ややこしいですが、こちらはOptionalです)
セットアップ
(1) Dockerの立ち上げ image file (Image: nvcr.io/nvidia/clara/bionemo-framework:1.5)
以下のコマンドはあくまで一例です。ご自身のGPU環境に合わせて、Dockerを立ち上げてください
#login
docker login nvcr.io
#Username: $oauthtoken
#Password <insert NGC API token here>
# Docker imageをpull
docker pull nvcr.io/nvidia/clara/bionemo-framework:1.5
# Launch Docker
docker run -it --rm --gpus all nvcr.io/nvidia/clara/bionemo-framework:1.5 bash(2) NGC CLI のダウンロード
# NGC CLI download
wget --content-disposition https://api.ngc.nvidia.com/v2/resources/nvidia/ngc-apps/ngc_cli/versions/3.42.0/files/ngccli_linux.zip -O ngccli_linux.zip && unzip ngccli_linux.zip
find ngc-cli/ -type f -exec md5sum {} + | LC_ALL=C sort | md5sum -c ngc-cli.md5
sha256sum ngccli_linux.zip
chmod u+x ngc-cli/ngc
echo "export PATH=\"\$PATH:$(pwd)/ngc-cli\"" >> ~/.bash_profile && source ~/.bash_profile
ngc config setAPI キーはNGCのAPIキーを入力してください
Org は ’no-org’ 以外のものを選択
format_typeは'json'を選択
その他はデフォルト値でOK
(3) 環境変数の設定
export BIONEMO_HOME="/workspace/bionemo"
export WANDB_API_KEY=<your WandB API key> # SaaSの場合は、https://wandb.ai/authorizeより取得可能です
# もしdedicated cloud版またはオンプレミス版のWandBを使用している場合は、"WANDB_BASE_URL"を適切に設定してください。
# あなたの会社のWandB管理者が"WANDB_BASE_URL"をご存知です。
[optional] export WANDB_BASE_URL=<your wandb base url>使用するcodeの準備
基本的にはBioNeMo Dockerイメージ内のsample scriptや関数を使用しますが、例題の一連の手順を入れたjupyter labや一部WandB連携のために変更する必要があるscriptをこちらのgithub repositoryに格納しています。手元にdownloadをして、ご自身の環境に配備してください(今後gitpullへの対応・そもそものDocker imageへの組み込みも考えております)。
ベースとしてはnvcr.io/nvidia/clara/bionemo-framework:1.5の中のデータとスクリプトを使いつつ、いくつかのスクリプトについては、https://github.com/olachinkei/BioNeMo_WandB/tree/mainの中のコードを使用します。
💡 Dockerのcustom imageの作成やgithubのrepositoryを今後用意しますが、updateが激しいBioNeMoなので、不格好ではありますがofficialのdocker imageに対してscriptを手動で足し算する形で進めます。
workspace 下に github repository内のProtein/01_protein_LLM.ipynb を配置し、このjupyter notebookをメインで使用します。
workspace/bionemo/examples/protein/esm2nv/pretrain.pyを github repository内のProtein/pretrain.pyに置き換えます。
workspace/bionemo/examples/protein/esm2nv/conf/base_config.yamlをgithub repository内のProtein/base_config.yaml に置き換えます。
01_protein_LLM.ipynbを開き、実行をしていきます。まずはWandBの環境変数の設定をし、WandB内で保存する場所を決定します。
import os
os.environ["WANDB_ENTITY"]="<your team where you want to log>"
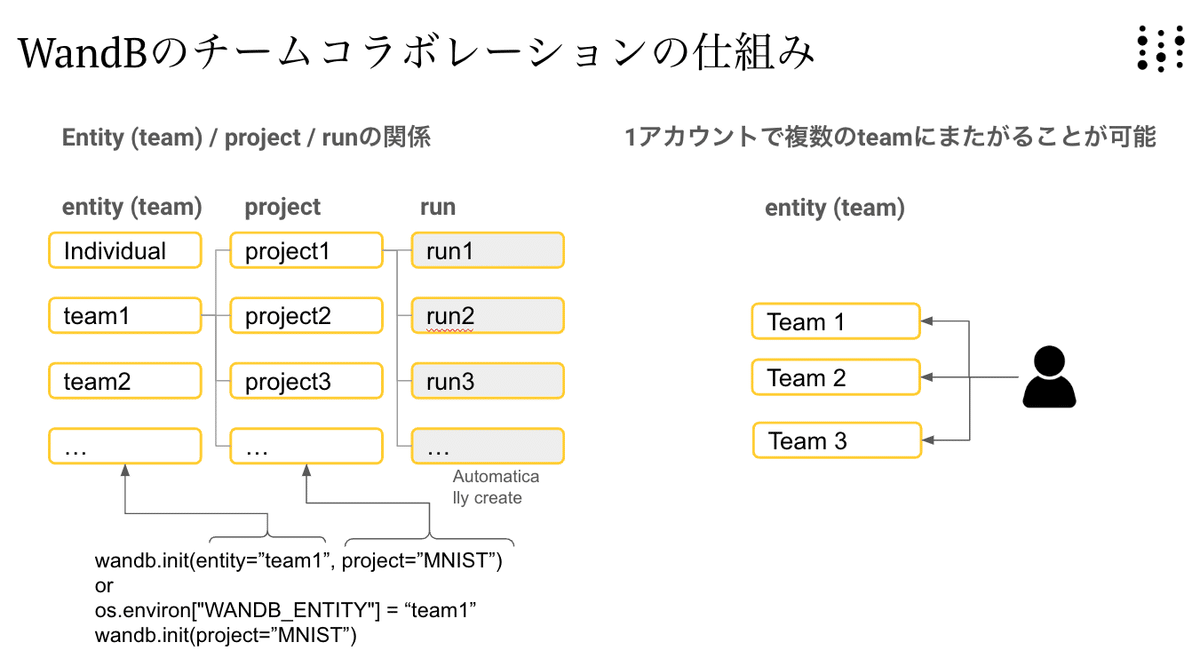
os.environ["WANDB_PROJECT"]="BioNeMo_protein_LLM_pretraining"WandBへの保存場所は、下図のような構成になっています。チームメートと同じentity (team)で作業をしていると、 自動的にリアルタイムにその結果が共有されます。
1. データ準備
環境構築ができたので、データ準備に進んでいきましょう。今回はBioNeMoのdocker imageに含まれているデータセットUniref50とUniref90を使用して事前学習を行います。
01_protein_LLM.ipynb内の"データの前処理"に進んでいきましょう。ここで、事前学習に使用するデータを準備しますが、今回はBioNeMoのdocker imageに含まれているデータセットUniref50とUniref90を使用します。
💡データについて詳しく知りたい方は"Language models of protein sequences at the scale of evolution enable accurate structure prediction"を参照してください。Uniref50 はトレーニング、検証、テスト用に分割されています。トレーニング中に Uniref50 のミニバッチがサンプリングされますが、このバッチの各シーケンスは、データのサイズと多様性を増加させるために、対応する Uniref90 クラスターのシーケンスに置き換えられています。
Uniref50は類似度が50%のシーケンスをクラスタリングして代表的なシーケンスを保存している一方、Uniref90は類似度が90%のシーケンスをクラスタリングして代表的なシーケンスを保存しています。つまりUniref90の方がより多くのデータが含まれているデータになります。Uniref90 はトレーニング時にのみ使用され、検証やテストには使用されません。
Data Prep 1
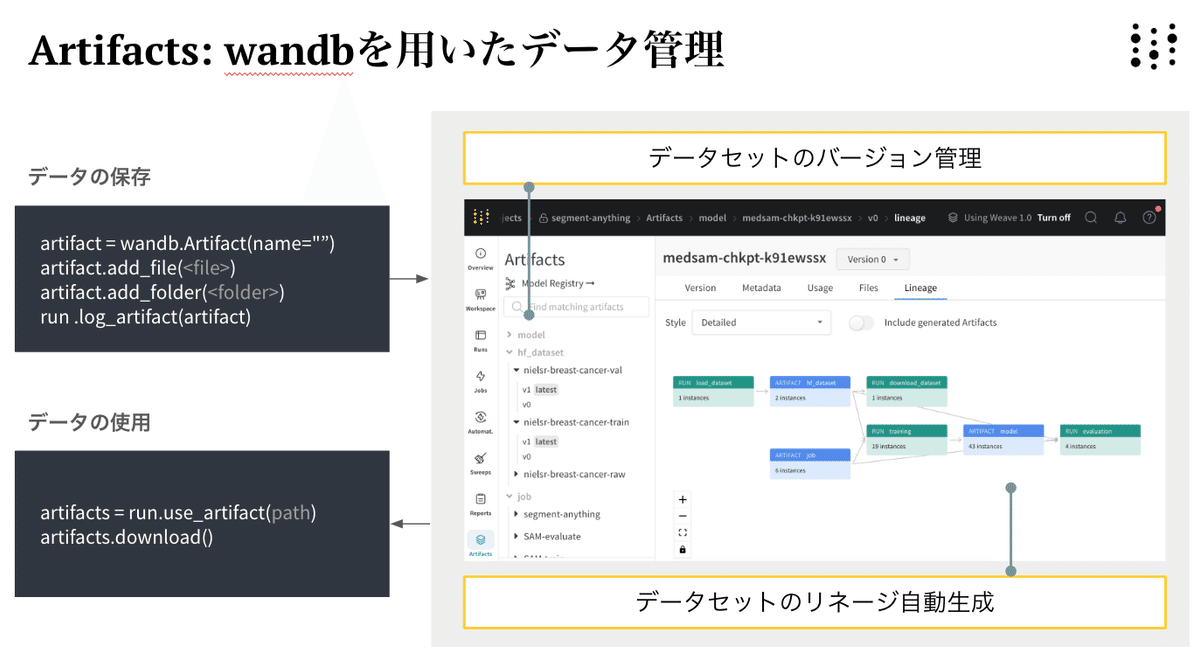
scriptを順番に実行し、uniref202104_esm2_qc_test200_val200.zipを解凍します。その後、データのバージョン管理のため、生データをWandBのArtifactsに保存します。Artifactsはデータやモデルなどのオブジェクトファイルのバージョン管理を可能にするツールです
Data Prep 2
次にデータの前処理を行います。以下のpretrain.pyでは、生データが入っているWandBのArtifactsを受け付け、再度別のArtfactsとしてWandBの登録するように変更しています。
!python examples/protein/esm2nv/pretrain.py\
--config-path=conf\
--config-name=pretrain_esm2_650M\
++do_training=False\
++exp_manager.wandb_logger_kwargs.name='preproceed_data_upload'\
++exp_manager.wandb_logger_kwargs.job_type='data_upload'\
++wandb_artifacts.wandb_use_artifact_path='${oc.env:WANDB_ENTITY}/${oc.env:WANDB_PROJECT}/uniref202104_esm2_qc_test200_val200:v0'\
++wandb_artifacts.wandb_log_artifact_name='uniref202104_esm2_qc_test200_val200_preprocessed'\
++model.data.val_size=500\
++model.data.test_size=100\
++model.data.uf50_datapath=/uniref50_train_filt.fasta\
++model.data.uf90_datapath=/ur90_ur50_sampler.fasta\
++model.data.cluster_mapping_tsv=/mapping.tsv\
++model.data.dataset_path=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uf50\
++model.data.uf90.uniref90_path=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uf90\
++model.data.train.uf50_datapath=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uniref50_train_filt.fasta\
++model.data.train.uf90_datapath=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/ur90_ur50_sampler.fasta\
++model.data.train.cluster_mapping_tsv=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/mapping.tsv\
++model.data.val.uf50_datapath=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uniref50_train_filt.fasta\
++model.data.test.uf50_datapath=/workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uniref50_train_filt.fasta
-- で始まるパラメータは、コマンドライン引数として pretrain.py に渡されます。例えば、 config-path と config-name は、設定 YAML ファイルのフォルダとyaml ファイル名を指定します。このパスは pretrain.py に対して相対的です。conf はexamples/protein/esm2nv/confを指し、pretrain_esm2_650M はexamples/protein/esm2nv/conf/pretrain_esm2_650M.yaml を指します。
++ で始まるパラメータは、YAMLファイルのデフォルト設定を上書きしたものとなっています。 base_config.yaml から継承された pretrain_esm2_650M.yaml の中のパラメータをいくつか説明します。
do_training: データの前処理のみを行い、トレーニングは行わないように False に設定します
exp_manager.wandb_logger_kwargs.name: wandbのrun nameを指定します
exp_manager.wandb_logger_kwargs.job_type: wandbのjob_typeを指定します(job_typeは後でrunの情報を整理する時に役立つメタデータになります。これによって実装が変わることはありません。)
wandb_artifacts.wandb_use_artifact_path: 前処理をするデータのwandbのartifactsのpathを設定します。
wandb_artifacts.wandb_log_artifact_name: 前処理をした後のデータを保存する際のwandbのartifacts名を指定します
処理が完了すると、前処理されたデータは /workspace/bionemo/examples/tests/test_data/uniref202104_esm2_qc_test200_val200/uf50/uf50/ に保存されます。もし独自のデータを利用して事前学習、ファインチューニングや推論をしたい場合は、パスを/workspace/bionemo/mydata/ に指定してください。ただし、データの構造やフォーマットをサンプルデータに揃える必要があります。
実行後、表示されるWandBのURLからWandBのダッシュボードにいき、Artifactsのページを確認しましょう。以下のようなリネージグラフが自動生成されていることがわかります。これでデータ準備の完成です。

事前学習
データの準備ができたので、その後のコードを順次実行し、最終的に以下のコードを実行して、事前学習を進めていきます。
!python examples/protein/esm2nv/pretrain.py \
--config-path=conf \
--config-name=pretrain_esm2_650M \
++do_training=True \
++do_testing=True \
++wandb_artifacts.wandb_use_artifact_path='${oc.env:WANDB_ENTITY}/${oc.env:WANDB_PROJECT}/uniref202104_esm2_qc_test200_val200_preprocessed:v0'\
++model.data.dataset_path=/ \
++model.data.uf90.uniref90_path=/uf90 \
++trainer.devices=1 \
++model.tensor_model_parallel_size=1 \
++model.micro_batch_size=4 \
++trainer.max_steps=1 \
++trainer.val_check_interval=1 \
++exp_manager.create_wandb_logger=True \
++exp_manager.checkpoint_callback_params.save_top_k=10以下、上記で設定している主なパラメータの説明です。
do_training: モデルをトレーニングするために True に設定します。これはデータが前処理されたことを前提としています。
do_testing: テストをスキップするために False に設定します。
wandb_artifacts.wandb_use_artifact_path: 学習に使用するデータセットのパスを入力します
trainer.devices: 使用するGPUの数を指定します。
model.tensor_model_parallel_size: テンソルモデル並列サイズを設定します。
model.micro_batch_size: バッチサイズを設定します。メモリエラーが発生しない限り、これをできるだけ増やします。
trainer.max_steps: トレーニングの最大ステップ数を指定します。デモのために 100 に設定しました。1 ステップ = 1 バッチの処理です。まず、total_batches = サンプル総数 / バッチサイズを計算します。N エポックでトレーニングしたい場合は、max_steps をN * total_batches に設定します。
exp_manager.create_wandb_logger: wandb へのログ記録を無効にするために False に設定します。True の場合は、WandB API キーを提供する必要があります。
なお、事前学習ではなく、継続事前学習を行いたい場合は、base_config.yamlの中のrestore_from_pathのスタートポイントとなるモデルのパスを入れることで行うことができます。
学習過程や設定はWandBに保存され、トレーニングされたモデルもWandBのartifactsに保存されます。以下は、WandBでトラックされた例です。
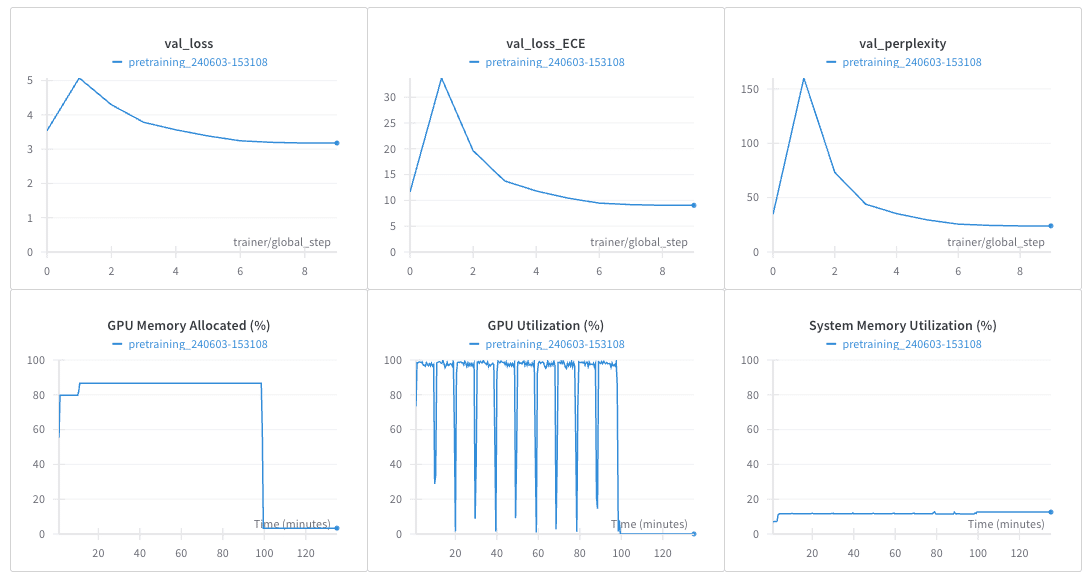

これで、タンパク質言語モデルの事前学習が完了しました。
Model Registryへの登録
Artifactsにモデルがあるままだと、他のチームの方がどのモデルが現在最高レベルのものなのか少しわかりづらい状態です。チームコラボレーションをより容易にするために、WandBのModel registryがあります。Model registryに事前学習したモデルを登録しましょう。手順は簡単です。登録したいモデルから"Link to registry"をクリックします。
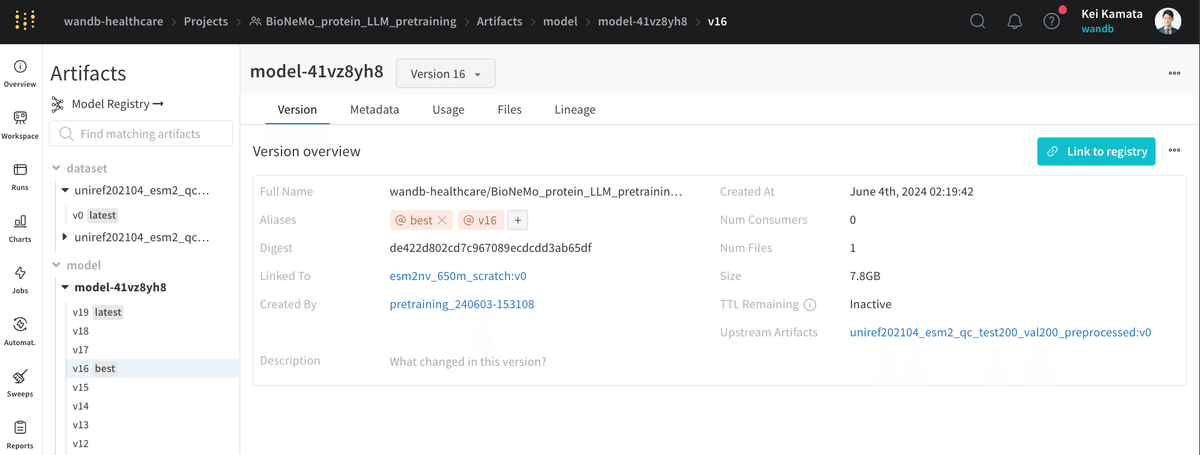
そうすると、組織のModel registryから今回のモデルが1つ管理されていることが確認できます。よりよい事前学習を行い、再度登録すると、自動的にv1, v2と新しいバージョンが作成されます。多くのバージョンができても、タグをつけることができるので、どのモデルがproductionで使われているのか、どのモデルがもっとも精度が良いかなどは簡単に管理することができます。

なお、Model Registryは2024年8月にupgradeしました。詳細は動画や公式ドキュメントをご覧下さい。
最後に
この記事では、BioNeMoを用いたタンパク質言語モデルの事前学習の実装例を紹介しました。Finetuningの例を確認されたい方は、"Pretraining & Finetuning of Protein Language Model with BioNeMO & WandB JP"をご確認ください。
実際には、データのクレンジングや準備は時間と労力がかかるところでありますが、モデルの学習自体は非常に簡単に行うことができることがわかっていただけたかと思います。分散学習もyamlファイルの設定を変更するだけで実現できるため、BioNeMoは使い勝手がよいフレームであるといえます。また、WandBを利用すると、学習のトラッキングをはじめ、データやモデルのバージョン管理も便利に行うことができることもお分かりいただけたかと思います。
多くの製薬企業様にハンズオンをする中で、「実装方法が今までわからず、心理的障壁が高かったが、一度やってみて前に踏み出せそう」という感想をいただくことが非常に多いです。こうした情報発信を引き続き行なっていければと考えています。
この記事の内容に関し、ご質問やフィードバックがありましたらcontact-jp@wandb.comまでご連絡ください。
この記事が気に入ったらサポートをしてみませんか?