【Aidemy X Bio】1222例の乳がんデータを深層学習させてみる!!(2) 遺伝子データを使ってRNN(深層学習)で癌診断してみた!

先日1222例の乳がんデータを可視化してみることを行った。詳しくは以下のノートを見ていただくとわかるが、
このデータには乳がん組織と正常組織の遺伝子発現情報が含まれている。今回このデータを用いて深層学習ができないか試みてみた。
このサンプルの遺伝子発現情報と、乳がんと正常のラベルを深層学習させ、そのあとでテストデータで正解率を確かめる、いわば癌の診断をさせてみようということだ!
RNNでがん診断!
学習モデルには時系列データや自然言語処理など一続き担ったデータを処理するのに有用なRNN、それを進化させたLSTM、GRUを比べてみた。モデルについてはいかが詳しい!
ますライブラリを読み込んで、RNNを試すこととする。
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import tensorflow
import keras
from keras.models import Sequential
from keras.layers import Dense, Activation, SimpleRNN
from keras import optimizersデータを読み込んで、成形して、形を確認
df_wine = pd.read_csv( 'breast.txt', sep="\t",header=0, index_col=0)
df_wine_2 = df_wine.T
df_wine_2 = pd.DataFrame(df_wine_2)
df_wine_2.shape
(1222, 13231)ここでわかるように、1222サンプルに対して、13231種類の遺伝子情報が載っている。
RNNの実装は
model = Sequential()
model.add(SimpleRNN(units, input_shape=x_train.shape[1:]))
model.add(Dense(y_train.shape[1], activation='softmax'))
のようになる。ユニット数は任意ではあるものの大体はデータポイント数(ここでは遺伝子数)と同じか同程度であるのが効率がよい。また予備実験でユニット数が1000を超えると計算にものすごく時間がかかることがわかっていた。
そこで分散が大きな遺伝子500に絞って学習を行わせることとする。
#遺伝子データ取り出し
X = df_wine_2.iloc[:, :].values
#分散(dispersion)計算
a = X.mean(axis=0)
b = X.var(axis=0)
c = b/a**2 - 1/a
c = pd.DataFrame(c)
c= c.T
#dispersionをデータにつけてソートし大きいものから順に500とった
Z = pd.DataFrame(X)
Z = Z.append(c, ignore_index=True)
Z.columns = df_wine_2.columns
Z.index = np.append(df_wine_2.index,'disp')
Z=Z.T
Z = Z.sort_values(by='disp', ascending = False)
Z=Z.T
X1 = Z.iloc[:1222, :500].values
ラベル情報を読み込んで, yに代入
t2 = pd.read_csv('type2.txt', sep="\t",header=0, index_col=False)
y = t2.iloc[:,0].valuesデータをRNNに使えるように、成形したのち、訓練データとテストデータに分離(*1)
from sklearn.model_selection import train_test_split
from keras.utils import to_categorical
x_train, x_test, y_train, y_test = train_test_split(x[:,:,np.newaxis], to_categorical(y), test_size=0.5)いよいよSimpleRNNで学習(活性化関数はSDGで勾配爆発予防のおまじないをclipvalu=1.で)
import tensorflow
import keras
from keras.models import Sequential
from keras.layers import Dense, Activation, SimpleRNN
from keras import optimizers
hid_dim = 500
model = Sequential()
model.add(SimpleRNN(hid_dim, input_shape=x_train.shape[1:]))
model.add(Dense(y_train.shape[1], activation='softmax'))
sgd2 = optimizers.SGD(lr=0.01, clipvalue=1.)
model.compile(loss='mean_squared_error', optimizer=sgd2, metrics=["accuracy"])
h=model.fit(x_train, y_train, epochs= 50, batch_size=20, verbose=1, validation_split=0.4)
score=model.evaluate(x_test,y_test, verbose=1)
print('Accuracy',score[1],"loss",score[0])
がん診断の結果
結果はブレブレの学習曲線だけれどもまあまあよい成績

テストデータでの成績も
Accuracy 0.988543371522 loss 0.0432175428137となかなかいい感じである!
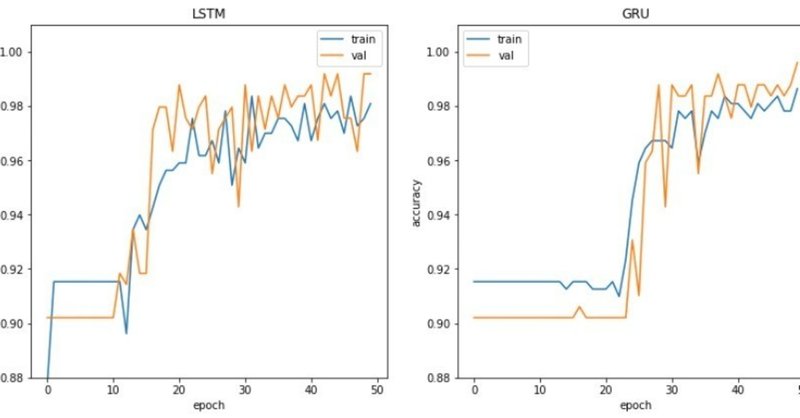
同様にLSTM、GRUでも学習を行ったが、simpleRNNよりは学習しにくいのか、うまく学習できていなかった。そこで活性化関数を変えて行ってみると
from keras.models import Sequential
from keras.layers import Dense, Activation, LSTM
hid_dim = 500
model = Sequential()
model.add(LSTM(hid_dim, input_shape=x_train.shape[1:]))
model.add(Dense(y_train.shape[1], activation='softmax'))
ada = optimizers.Adagrad(lr=0.001, clipnorm=1.)
model.compile(loss='categorical_crossentropy', optimizer=ada, metrics=["accuracy"])
h2=model.fit(x_train, y_train, epochs= 50, batch_size=20, verbose=1, validation_split=0.4)
score=model.evaluate(x_test,y_test, verbose=1)
print('Accuracy',score[1],"loss",score[0])
でテストデータでの評価も
Accuracy 0.988543371522 loss 0.0432175428137と悪くない感じ
GRUも同様に
from keras.models import Sequential
from keras.layers import Dense, Activation, LSTM, GRU
hid_dim=500
model = Sequential()
model.add(GRU(hid_dim, input_shape=x_train.shape[1:], activation='tanh', recurrent_activation='hard_sigmoid', use_bias=True,
))
model.add(Dense(y_train.shape[1], activation='softmax'))
ada = optimizers.Adagrad(lr=0.001, clipnorm=1.)
model.compile(loss='categorical_crossentropy', optimizer=ada, metrics=["accuracy"])
h3=model.fit(x_train, y_train, epochs=50, batch_size=20, verbose=1,validation_split=0.4)
score=model.evaluate(x_test,y_test, verbose=1)
print('Accuracy',score[1],"loss",score[0])と指定し

テストデータでの評価も
Accuracy 0.981996726678 loss 0.0457487605777と同様でした。
ちなみにこの活性化関数の方が学習を起こしやすいのかsimpleRNNでやってみると最初から学習が飽和している感じでした

テストデータの評価も
Accuracy 0.981996726678 loss 0.093449141081といった模様でした。最後にもう一度3つのモデルを比較すると
import matplotlib.pyplot as plt
plt.figure(figsize=(20, 6))
plt.subplot(131)
plt.plot(h.history["acc"])
plt.plot(h.history["val_acc"])
plt.title("SimpleRNN")
plt.legend(['train',"val"])
plt.xlabel("epoch")
plt.ylabel("accuracy")
plt.ylim(0.88,1.01)
plt.show
plt.subplot(132)
plt.plot(h2.history["acc"])
plt.plot(h2.history["val_acc"])
plt.title("LSTM")
plt.legend(['train',"val"])
plt.xlabel("epoch")
plt.ylabel("accuracy")
plt.ylim(0.88,1.01)
plt.show
plt.subplot(133)
plt.plot(h3.history["acc"])
plt.plot(h3.history["val_acc"])
plt.title("GRU")
plt.legend(['train',"val"])
plt.xlabel("epoch")
plt.ylabel("accuracy")
plt.ylim(0.88,1.01)
plt.show
これだと、SimpleRNNが一番よいのでしょうか?いいのだろうか笑
今後の課題
不均衡データへの対策:今回使ったサンプルでは98%程度とよい精度で癌を診断できた。このサンプルの場合10%程度が正常、90%程度が癌である。すこし不均衡なデータであり、じつはトレーニングデータの取り方によっては正常があまり含まれておらず、意味ある学習ができなくなることがある。じつは予備実験で色々なデータをつかったのであるが、1222例のサンプルのうち、300例をつかった学習ではうまく学習できなかった。
にあるように、
識別問題において,各クラスのデータが生じる確率に大きな差がある場合.例えば,二値識別問題で正例が 1% で,負例が 99% といった状況.はずれ値検出を識別問題として解く場合などが該当する.こうしたデータについては,予測精度が非常に低下する場合があることが知られている.
不均衡データについて対策すればもう少し小さなサンプルでも対応できるようになるのかもしれない。これついては、以下のサイトにあるように、SMOTE(synthetic minority over-sampling)などの手法で少ないクラスのデータをオーバサンプリングする都行った手法が開発されており、pythonのライブラリも用意されているので、今後こういった手法を導入して試してみたい。
遺伝子データをRNNで機械学習することの妥当性: 遺伝子データは多種類であるものの、一次元のデータで時系列データに似ておりそのアナロジーからRNNを使用してみた(今週DL4USでRNNがテーマだったこともある)。一方でRNNでは多数のユニットによる学習には計算時間を要するため、dispersionの大きい(つまり一番差の見やすい)遺伝子500に絞って学習を行っており、一部の情報を捨てての学習になる。今回のように正常、異常の2群の比較であれば問題ないと思われるが、3群以上の分別の場合は、小さな差を捨てるこの方法は難があるかもしれない。前もってPCAやtSNEなどで次元を削減して学習させてみるといった手法も考慮した方がいいかもしれない。
また遺伝子データをRNNで機械学習するといったテーマであまり検索が引っかかってこない。あまり使われていないということは遺伝子データに対しては別のよい手法があるのかもしれない。もしどなたかよい方法をご存知であれば教えて欲しい!
活性化関数の選択:今回検討した中では活性化関数の選び方によって学習が全くしない事例があった。理由がいまひとつはっきりしないのと、ベストな活性化関数が選べているかどうか不明なため、今後探っていきたい。
一次元畳み込みネットワークの可能性:時系列データを用いた予備的な解析ではCNNの方が成績がよい事例もあった。CNN vs RNNについても今後探っていきたい。
注1:遺伝子データにチャンネル数の次元を加えること: x[:,:,np.newaxis], ラベルデータをカテゴリかすること: to_categorical(y)が重要である。またデータが不均衡であるため、できるだけ含まれる正常細胞の割合が偏らないように、訓練データとテストデータは1:1に分けた。
この記事が気に入ったらサポートをしてみませんか?