バイオインフォマティクスのすすめ(2)

第2回目の今回は,前回取得した配列情報を用いて,実際に配列アライメントを行っていきます.
目次
0.始めに
1.Network Protein Sequence @nalysis
2.Clustal W の利用と結果表示
3.ESPriptを用いた出力方法
4.今回のアライメントに対する考察
5.まとめ
0.始めに
配列アライメントとは,比較したい複数(n ≥ 2)の配列を整列させて,その配列中のどの文字が一致し,どの文字が不一致で,どの領域が欠失 or 挿入されているのかを検索する手法です.
ここで注意しなければならないのは,ここで得られる情報は完全に正しいわけではないということです.
以下に例を2つ示します.この2例は立場が逆転しています.比較・想像してみましょう.特に(例2)は,極論ではありますが…
例1)タンパク質Aの機能が明らかになっているとします.この相同(同一起源を持ち,進化的に近い)タンパク質Bをターゲットとして扱う場合,Aの活性中心(機能的に重要な領域)が既知なので,配列アライメントの結果からBの活性中心を予測することができます.しかし,これは,あくまで予測なので,その活性中心の予測領域が本当に機能するかは,実際に実験で明らかにする必要があります.
例2)もし,配列アライメントという手法を知らなければ,AがBの相同タンパク質であることを知ることができません.その結果,Aの活性中心が分かっているのに,Bの活性中心を予測できず,N〜C末端まで順に(もしくはランダムに)変異体を作製して活性測定を行い続けなくてはなりません.
立ち位置が補助的といえども,配列アライメントに限らずバイオインフォマティクスの各手法を用いることで,生物系・生命科学系の実験ユーザーの仕事量が少なくて済みます.仕事が減ることによって,視野を拡げることができ,空き時間を有効利用することも可能になるかと思います.
正直,この現代に,例2に該当する研究者はいないのではないかな?とは思いますが…
1.Network Protein Sequence @nalysis
配列アライメントは,実は色んなサーバーで行うことができます.それぞれのサーバー(サイト)に癖・特徴があって,どれも一長一短だと思います.
今回は,配列解析コンプレックスサイトであるNetwork Protein Sequence @nalysis (NPS@) を用います.
それでは早速 NPS@ に行ってみましょう.
Googleで NPS@ と検索するか,以下のURLをコピペしましょう.
https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_server.html
Googleで検索すると上から2番目あたりにリンクが出てきます.

Network Protein Sequence @nalysis
このサイトは名前の通り,遺伝子(タンパク質,核酸とも)の配列を解析するソフトウェア(サーバー)の集合サイトです.前回,FASTAのところで登場したBLASTもあります.BLASTについては,それだけで特集を組んで説明する予定です.
今回は,Multiple alignment の "Clustal W Protein sequences" と "Clustal W DNA sequences" を用います(下図).

2.Clustal W の利用と結果表示
それでは,NPS@ → Multiple alignment → Clustal W DNA sequences を開いてみましょう.

上図の通り,FASTA形式で整列させた核酸配列(2本以上)をクエリーボックス(白い背景の場所)に貼り付けます.今回は,前回取得した,bsu_ftszとsau_ftszのDNA配列を用います.
貼り付けた配列が,FASTA形式であることと核酸配列であることをダブルチェックし,SUBMITボタンをクリックします.SUBMITの下に幾つかオプションがありますが,特に気にしなくて大丈夫です.
配列アライメントの結果は以下のように表示されます.

bsu_ftszとsau_ftszの配列を比較した結果,赤字で表示される塩基(例えば先頭のATGTT)は2本の配列間で「一致」していることを示しています.一方,黒字の塩基は2本の配列間で「不一致」であることを示しています.ここで示した配列アライメントは一部ですが,実際のアライメント(上図)の他,データサマリーもアライメント結果の下方に表示されます(下図).

次に示すアミノ酸配列の配列アライメントでも同じサマリーが表示されますので,ここで説明しておきます.
配列アライメントは,もちろん具体的な配列の差を見ることができるツールであるとともに,全体的にどれくらい同じで,どれくらい異なるのかを%で指標できるツールでもあります.それが上図の3〜6行目にあたります. "Identity" は「一致度」, "Strong similar" は「高い類似度」, "Weakly similar" は「低い類似度」, "Different" は「不一致度」を示しています.
DNA配列はその遺伝子(発現するとタンパク質)を決定する最高位に位置しています.セントラルドグマの矢印を思い浮かべると早いですね(下コラム).
DNA → RNA → タンパク質
DNA配列の場合,DNA配列を決定づける上位の配列が存在しないため,配列比較を行うと,両者が一致する(Identity)かしない(Different)かという結果しか得られません.そのため,上図のとおり,Strong similar や Weakly similar は 0% となります.今回のアライメントのIdentityは ~65%,Differentは ~35% です.
次に,アミノ酸配列の場合は,NPS@ のトップページの Clustal W Protein sequence(下図)を選択します.その先は核酸配列のアライメントの時と同じです.

アミノ酸配列の配列アライメントの結果を表示します.
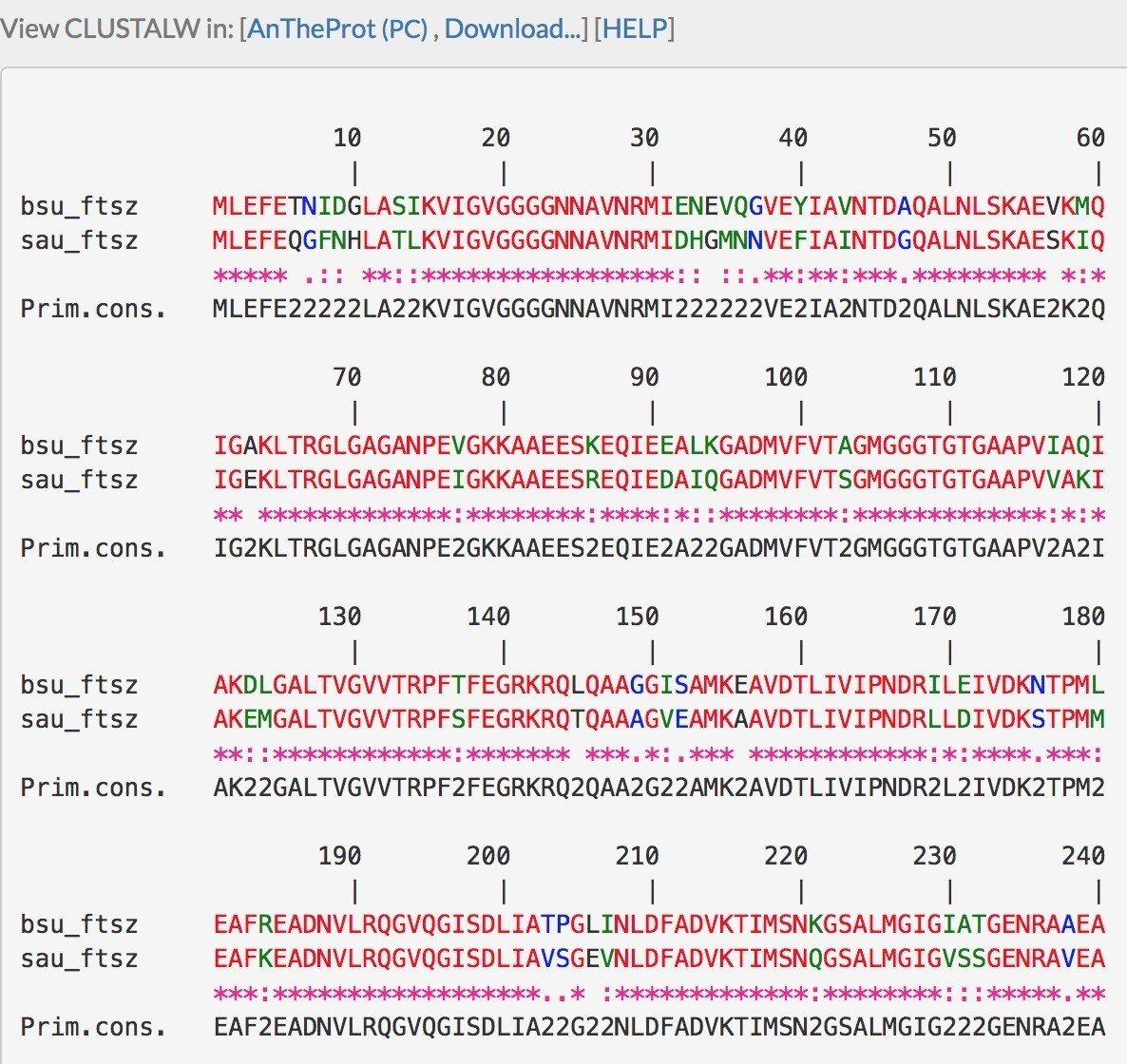
DNA配列の配列アライメントと同様に,アミノ酸配列では,赤字の残基は「一致」を,黒字の残基は「不一致」を示しますが,アミノ酸配列の場合,緑字と青字があります.緑字は Strongly similar を,青字は Weakly similar を示しています.

先述の通り,アミノ酸配列の上位にDNA配列があるため,2つの生物種間でアミノ酸が異なる場合,それは両者のDNA配列が異なることになります.そのため,Strongly similar や Weakly similar が存在します.Similar のみならずStrongly や Weakly が存在するのは,進化の過程で起こる突然変異の起こりやすさから来ています.
上図のとおり,当然のことですが,同じタンパク質の同じ生物種の比較でもDNA配列とアミノ酸配列では類似度が異なります.
3.ESPriptを用いた出力方法
ESPript は NPS@ の Clustal W で出力した配列アライメントの結果に色づけをして見やすくするソフトウェアです.
数年前に NPS@ がリニューアルされてから,前のセクションで示した配列アライメントの出力結果でも充分に扱いやすくなったおかげで,ESPript を利用する機会が無くなってしまったのですが,良い機会なので紹介しておきます.
まず,アミノ酸の配列アライメントの結果表示のページで,ESPript のボタンを探しましょう(下図).

ここをクリックすると,ESPript のページに行きます(下図).

NPS@ から,このページに飛ぶと,NPS@ の Clustal W で表示した配列アライメントの結果がそのまま引き継がれます.時間が経過しすぎるとリンク切れになることもあるので注意してください.
また,この ESPript を利用する上で注意しなければならないのは,ほぼ必ずポップアップブロックされるということです.サイトを信用して利用して頂けると嬉しいです(開発者ではないですが).
① 上部タブの "MODE" を "BEG" から "ADV" に変更します.

すると最上部の "Aligned Sequence" のコラムがさらに詳細に設定することができるようになります.また2番目のコラムで,配列アライメントの結果に二次構造(その配列の構造情報が既知の場合)を加えることもできます.
② 配列アライメントをきれいに表示させるために各種設定をします.

デフォルトでは上図のように,"Display consensus seq." にチェックが入っているので,これを外します.
また,"Similarity coloring scheme" では配列の一致度,類似度をどのアルゴリズムに沿った色分けにするのかを選択できます.配列がどれだけ似ているか以外に,どれだけ進化的に近いのかも見たい場合は,PAM や BLOSUM が推奨されますが,ここでは,Clustal W の結果を見やすく表示するだけなので,デフォルトの "%MultAlin" で問題ないと思います.しかし "%MultAlin" から,後々,Adove Illustrator などを用いて,ユーザーの手で変更したい場合に少々厄介ではあります.
③ 出力レイアウトを設定します.

日本で研究する場合は,Paper size は A4 で良いでしょうね.アメリカなら US letter にしておきましょう.デフォルトでは Font size が "5" になっていて,これは小さすぎるので "12" くらいに変更すると良いと思います.

また,出力ファイル形式も設定できます.後々のために,デフォルトの PostScript file と PDF file を選択します.
④ 出力します.

上部タブの "SUBMIT" をクリックすると,おそらく最初はうまく行かないです.それはポップアップブロックされるからなのでブロックをオフにしましょう.その後,お手数ですが,NPS@ の Clustal W から ESPript を開く操作からやり直してください.
(かく言う私もこのブログを書きながら実際に操作を進めたら,しっかりとポップアップブロックされ,今,やり直しているところです)
⑤ 結果表示
ポップアップブロックを解除して出力に成功すると,上図のような結果ページが表示されます.RESULTS のタブの PDF file の pdf [11 Kb] をクリックすれば PDF ファイルが開きます.

上図はこのページに貼り付けるために PNG で出力したものですが,PDF でも同様に表示されます.赤い背景は「一致」を示し,黄色い背景は「類似」を,一部のドット( . . . )は「挿入 "Insert" ・欠失 "deletion" (合わせて "Indel" と呼びます)」を示します.
これで,ESPript を用いた配列アライメントのレイアウトは完成です.
4.今回のアライメントに対する私見
今回はBacillus subtilis(枯草菌) と Staphylococcus aureus(黄色ブドウ球菌)の FtsZ という細胞分裂に関与するタンパク質のアミノ酸配列とDNA配列の配列アライメントを行ったので,その比較と簡単な私見で終わりたいと思います.
枯草菌も黄色ブドウ球菌も同じバクテリアです.実際に明らかにされたFtsZの結晶構造からもそこまで大きな差はないと知られています.しかし,配列アライメントの結果(上図)から,N末端領域からの8割程度の配列は,確かに両者間で大きな違いがないものの,C末端領域では少し差が見られます.配列が異なるのは進化の過程での突然変異や環境適応のためであることは真実です.それを明らかにすることは,そのタンパク質の機能を明らかにする上で重要です.しかし,それ以上に,これらの違いの正確な解明が,感染症に対する特効薬の開発や新規農薬ターゲットなど,我々の身の回りの生活でも役に立つということを押さえて頂けると幸いです(実際に既にターゲットになってる気もしますが…)
5.まとめ
今回もしっかり長くなってしまいましたが,配列アライメントの出力とよりきれいに表示させるためのソフトウェアの使い方の基本を解説しました.もし今後,使ってみたいと思ったときの役に立てると良いなと思っています.
次回は,配列アライメントの中について解説していきます.自分はプログラム屋ではないので,あまり突っ込んだ説明はできないですが…
今後ともどうぞよろしくお願いいたします.
この記事が気に入ったらサポートをしてみませんか?