バイオインフォマティクスのすすめ(5)

長らく更新できませんでしたが,第5回目の今回は,前回解説したローカル・アライメントを用いた遺伝子配列の相同検索について説明していきます.
0.始めに
1.遺伝子を見つける
2.BLAST とは
3.BLAST を使ってみよう
4.BLAST のアルゴリズム
5.BLAST の評価方法
6.まとめ
0.始めに
ある遺伝子(タンパク質や RNA を発現する DNA 配列)の情報を見つけたいときどうしますか?ある DNA 配列を持っているときに,それが何かを同定したい場合は何をしたら良いでしょうか?
以下に示す図は,Human の eEF-2 の DNA を示しています.しかし,真核生物の核内で起こる特徴的なパスウェイ「スプライシング」を考慮していないため,実際の配列数:2895 塩基に対して,ここで示す配列数は 3163 塩基です.これを眺めているだけでは,どの 2895 塩基が将来,eEF-2 の mRNA になるのか分かりません.

1.遺伝子を見つける
DNA 配列が何のタンパク質をコードしているのか,つまり,将来的に何というタンパク質になるのか,が理解できなければ,そのDNA 配列はただの文字列で何の価値もありません.
配列を同定するためには,幾つかの方法が知られています.1 つは遺伝子予測ソフトを使う方法です.もう1 つは,既に知られている遺伝子配列との配列比較により相同検索する方法です.
今回は,2 番目の方法を取り上げて解説していきます.
2.BLAST とは
ある1 本の配列と類似する配列がデータベース中にあるかを調べるために, BLAST や FASTA などのアプリケーションを用います.FASTA は FASTA 形式に登場したアレです.しかし現在ではほとんどの利用者が BLAST を用います.そのため,BLAST は世界標準の配列相同検索アプリケーションとして君臨しています.
BLAST は,Basic Local Alignment Search Tools の頭文字を取った略語です.勘の良い方はお気づきかもしれませんが,前回の第4回の後半で紹介した Local Alignment を用いた計算ソフトです.Local Alignment のメリットは,1本の配列を断片化することで,その断片と比較配列がどれだけ一致するのか?類似するのかを見ることでした.それゆえ BLAST では,断片化したクエリー配列を配列データベース上に投げて検索をかけることで,まず一致もしくは類似配列を見つけます.そのうえで,その断片配列を伸長させていき比較配列との近さを評価します.
BLAST には,用途に応じて幾つかの種類があります.一番分かりやすいのは配列の種類です.さすがに,アミノ酸配列と DNA 配列を直接比較することはできません.理由は用いる文字が異なるからです.以下に BLAST の種類を示します.
1.blastn
塩基配列をクエリー配列として,塩基配列データベースから相同配列を検索する方法
2.blastp
アミノ酸配列をクエリー配列として,アミノ酸配列データベースから相同配列を検索する方法
3.blastx
塩基配列をクエリー配列として,それを 6 フレーム分アミノ酸配列に翻訳し,アミノ酸配列データベースから相同配列を検索する方法
4.tblastn
アミノ酸配列をクエリー配列として,塩基配列データベースを 6 フレーム分アミノ酸配列に翻訳しながら相同配列を検索する方法
5.tblastx
塩基配列をクエリー配列として,これを 6 フレーム分アミノ酸配列に翻訳しつつ,塩基配列データベースも 6 フレーム分アミノ酸配列に翻訳しながら相同配列を検索する方法
3.BLAST を使ってみよう
これまでも何度か説明したように,BLAST とは世界標準のローカル・アライメント検索ツールです.「ツール・アプリケーション」は利用場所を選びません.そのため,塩基配列・アミノ酸配列データベースを管理しているサイトのほとんどには BLAST が導入されています.そのため,どのサイトからBLASTを用いても問題ありません.しかし,当たり前のように聞こえるかもしれませんが,BLAST の検索結果は用いたデータベースに依存します.
では早速 BLAST 検索をしていきましょう.ここでは,KEGG GENES から行います.下図の通り,AA seq ボタンをクリックして表示された配列情報の下にある BLAST ボタンをクリックします.オプションを変更したい場合はそれぞれ選択して,Compute をクリックします.

このKEGG GENES の場合は,検索データベースとして NCBI GenBank を用いています.以下に結果を表示します.

まず,BLAST の結果は上図左に示されます.クエリー配列に対する一致度・類似度の高いタンパク質の Entry ID とタンパク質の慣用名が表示され,右側には bits や E-value といった類似(相違)度を表す統計値が記されます.
今回のクエリー配列は BLAST サーチには向いておらず非常に悪い例で申し訳ないです.
ご存じの通り,Staphylococcus aureus(黄色ブドウ球菌)は知られている株だけでも相当数ある(今後も増え続ける)ため,クエリー配列と同じ配列(および末端欠損体も含む)を持つ FtsZ が沢山あることになります.そのため,E-value の値が 0(後述しますが,0 は完全一致を示します)になっています.
そのため,Top 10 を選択すると全て同じタンパク質(菌の株は違う)と比較することになるため,自分で画面をスクロールしていき,適当に 10 種類程度選択します.
そして「Select operation」で表示したい項目を選択します.上図では TREE と CLUSTAL W を紹介しています.TREE とは名前が示す通り,進化系統樹です.この場合,比較した FtsZ が互いにどれだけ近いのかを示しています.Clustal W については説明は不要ですね.この「バイオインフォマティクスのすすめ」シリーズの(2)で紹介しました.
4.BLAST のアルゴリズム
次に,BLAST がどういう仕組みで検索を行っているのかについて簡単に紹介します.
① クエリー配列を用意します.この配列は必ず 1 本鎖である必要があります.ただし入力時における画面上のスペース・改行は問題ありません.
② クエリー配列を断片化して,文字列のリストを作成します.
③ これらの文字列を用いて配列データベース検索を行います.
④ 実際に配列アライメントを行い,一致度の高い配列をピックアップしていきます.
⑤ 一致度の高い配列を見つけたら,文字列の両端を伸ばしていき,最終的に全長での配列アライメントを出力します.

5.BLAST の評価方法
「3.BLAST を使ってみよう」で,検索結果に bits や E-val が登場しましたが,あれが BLAST の評価です.BLAST には,発見した配列に対して Identity, Sore (Bit), E-value が出力されます.どうして複数の評価方法があるかというと,これは開発者と末端利用者の専門性の違いにあると思われます.開発者(計算屋)が考える類似性の評価基準が必ずしも末端利用者の利用目的に準ずるとは限らないということです.そのため複数の評価方法を用意して,利用者の好む(目的に合う)統計値を選択する幅を設けているわけです.
では,Identity, Sore (Bit), E-value について,1つずつ紹介していきます.
① Identity
Identity とは名前の通り,クエリー配列に対してサブジェクト配列がどれだけ似ているのかの比率(%)を示します.
以下は Sau_FtsZ をクエリー配列としたときの 2 種類のサブジェクト配列との配列比較における Identity の例です.
2種類のサブジェクト配列:
sams:NI36_05755 cell division protein FtsZ
sje:AAV35_007450 cell division protein FtsZ
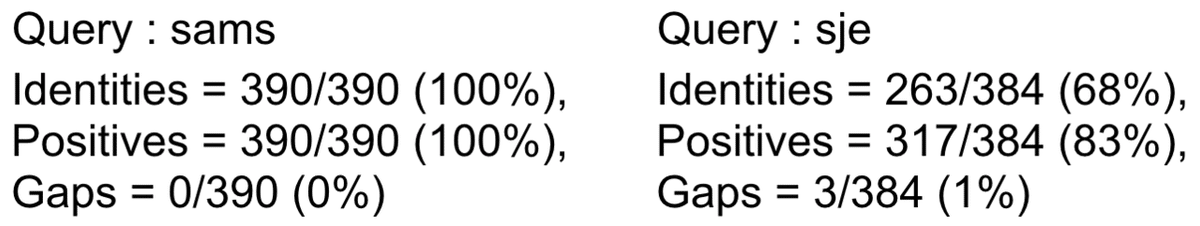
sams はクエリー配列とほぼ同じことが分かります(同じ生物種の別株).Sau_FtsZ は全長が 390 残基あるので,sams は完全一致しています.
それに対し,sje では分母が 384 残基と 6 残基少なくなっています.これはsje の N もしくは C 末端または両端が計 6 残基分短いことを示しています.sje の場合,一致する文字が 263 残基,一致+類似性の高い文字の合計が 317 残基,ギャップ(Indel)が 3 残基分あることを示しています.
この評価方法 Identity の問題点は,直感的で感度が悪く,注目している 2 本の配列の相違点しか分からないところです.もちろん直感的というのは利点でもあります.
② Score と Bit score
Score とは,置換行列により計算される類似度の尺度で,Identity よりも感度が高いものの比較する配列に依存します.これを改良したのが,Bit score です.Bit score は Score を正規化したものなので,配列長やデータベースに依存しません.以下の式は Score を示しています.

3 つある評価方法の中でも,Score は唯一利用したことがないので,これ以上の説明が出来ずに申し訳ないです.今後,Score と遭遇する機会があればもう少し掘り下げて勉強してみようと思います.実際に,Identity で利用した2種類のサブジェクト配列との比較における Score を以下に表示します.

Score が低い方が類似性が低いことが分かります.
③ E-value
E-value とは,期待値のことです.E は Expected の略です.
ランダムな配列データベースを検索したときに,そのスコア S 以上の値になるアライメントの本数の期待値を表示しています.
ただ説明を書いてもよく分からないですよね.つまり,
論理展開(① → ② → ③)
① ランダムな配列では起こりえないスコア
② 偶然では起こりえないスコア
③ 進化的に関係がある類似性に違いない
ということです.
そもそも,期待値とは高校数学の確率論で習いますが,せっかくなので復習してみましょう.
<期待値>
1回の試行で得られる値の平均
1回の試行で獲得しうる全ての値(サイコロの目なら 1 〜 6 の数字)とその値が出る確率(サイコロならそれぞれ 1/6)の積を足し合わせて得られる.
E = 1 * (1/6) + 2 * (1/6) + 3 * (1/6) + 4 * (1/6) + 5 * (1/6) + 6 * (1/6)
= 3.5
となります.
獲得する値は設定により変化するので,今回はサイコロの目にしましたが,金銭にもユーザー設定の値にも変更可です. 対象によっては確率が一律ではない場合も当然あるでしょう.
話を元に戻します.この期待値の計算では,単位は「本」です.アライメントの本数ですね.なので,必ず0以上の数値になり,値が小さいほどよく似ていることになります.
E-value の式は以下の通りです.

K と λ はスコア行列とギャップペナルティに依存するパラメータ定数です.
m はクエリー配列の残基数で,n はデータベース中の配列の総残基数です.つまり,データベースに登録されている配列を一列に並べたときの長さです.
上式の通り,E-value はクエリー配列長(m)とデータベース配列長(n)に比例します.これはクエリー配列が同じでも用いるデータベースによって値が変化することを示しています.
そのため,必ず注意しなければならないことがあります.例えば,A と B の2 つの配列を比較したいときに,それぞれ別々のデータベースの BLAST で検索をかけた上で,A の方が E-value が低く似ているという判断を下してはいけません.
6.まとめ
1 月上旬に私用(学会)があり,その後は実験で忙しくしていたため,この第5回目を仕上げる時間が取れませんでしたが,いかがでしたでしょうか.
本編でも述べていますが,BLAST を初めて利用すると,上司や先輩によってどの統計値を優先して見るべきかが異なります.自分ももちろんその一人で,自分の場合は上司が E-value 派だったので,当然,後輩に指導するときは E-value を押しているような気がします.
第4回のまとめにも書きましたが,自然界における進化をどうにかして数値化・統計化している時点で,そこに不完全性は付きものです.我々,末端ユーザーはそれらを許容しつつ,複数ある統計値を自分の用途に合わせて用いるべきだと思います.
次回は遺伝子を見つける方法のうち,遺伝子予測ソフトを用いる場合の一般例を紹介しようと思います.自分は専門がもっと下流のタンパク質やRNAの構造生物学なので,自ら遺伝子の同定などは行ったことがなく,知識が追いついていない感じもしますが,そこはご承知頂きたく思います.
それでは,今後ともどうぞよろしくお願いいたします.
この記事が気に入ったらサポートをしてみませんか?