ヒト組織のマイクロバイオーム解析に最適化された細菌DNA分離法

記事内容へスキップ
記事情報へスキップ
ワイリーオンラインライブラリー
微生物学オープン第10巻第3号e1191
原著論文
オープンアクセス
ヒト組織のマイクロバイオーム解析に最適化された細菌DNA分離法
https://onlinelibrary.wiley.com/doi/10.1002/mbo3.1191
カルライン・E・ブルッゲリング、ダニエル・R・ガルザ、スミア・アチュイティ、ワウテル・メス、バス・E・デュティル、アンネマリー・ボレイ
https://doi.org/10.1002/mbo3.1191
引用 11
について
セクション
図解抄録
ヒト大腸生検の細菌DNA分離法を紹介する。この分離法では、ヒトのDNAを減少させ、門レベルでの相対的細菌存在量を歪めることはない。ショットガンシーケンスの結果、508/521のメタゲノムが得られた(Shannon index; 2.9, UniFrac distance; 0.56)。15個の結腸生検について、アンプリコンシーケンスとショットガンシーケンスの両方で塩基配列を決定した結果、門レベルから属レベルまで同様の細菌プロファイルが得られた。このプロトコールにより、臨床組織サンプルの幅広い細菌スペクトルの解析が容易になり、マイクロバイオーム研究への応用性が向上した。
詳細不明
概要
近年のマイクロバイオーム解析の進歩により、ヒトの健康におけるマイクロバイオームの役割について新たな知見が得られ、臨床的意義が期待されている。残念ながら、組織分離株には宿主DNAが存在するため、宿主関連細菌の解析は妨げられてきた。ここでは、切除したヒト大腸からの生検(大きさ2~5mm)に最適化した、組織用のDNA分離プロトコルを紹介する。このプロトコールでは、門レベルでの相対的な細菌存在量を歪めることなく、ヒトDNAを減少させることができる。どの濃度のトリトンとサポニンがヒト細胞を溶解し、細菌細胞を無傷のまま残すかを、放出されたヒトDNAを枯渇させるDNAse処理と組み合わせて評価した。サポニンはPBS中0.0125%の濃度で宿主細胞を溶解し、qPCRで評価したファーミキューテス、バクテロイデーテス、γ-プロテオバクテリア、および放線菌の相対量を維持しながら、細菌DNAを4.5倍に濃縮した。われわれの最適化されたプロトコルは、ショットガンメタゲノミクスを用いて、226人の患者の521のin vivo大腸生検を対象とした2つの大規模臨床研究において検証された。その結果、得られた細菌プロファイルは、16S rRNAアンプリコンシークエンシングによって見出された多様性に類似したαおよびβ多様性を示した。15例の鉗子組織生検のショットガンメタゲノミクスと16S rRNAアンプリコンシーケンスを直接比較した結果、両シーケンス間で類似した細菌プロファイルと類似したシャノン多様性指標が示された。これにより、すべての細菌を効率的に分離できる、組織生検からの細菌DNA濃縮のための最初のプロトコルを提示する。このプロトコールにより、臨床組織サンプルの幅広い細菌スペクトルの解析が容易になり、マイクロバイオーム研究への応用性が向上する。
1 はじめに
急速に発展しているマイクロバイオーム研究の分野では、ヒトの健康や疾患におけるマイクロバイオームの役割が着実に明らかにされつつある。腸内マイクロバイオームの機能は多様であり、代謝、組織の恒常性、免疫に関わる多くの生物学的プロセスに不可欠である(Lynch & Pedersen, 2016)。マイクロバイオーム組成の変化は、腸炎症性疾患から大腸がん、消化管以外の疾患に至るまで、多種多様な疾患と関連している(Lynch & Pedersen, 2016)。このような組成の変化は、分離したDNAの塩基配列決定によるマイクロバイオーム・プロファイリングによってよく研究されている。これまで膨大な量の研究が便を用いて行われてきたが、最近の技術により大腸組織での細菌プロファイリングが容易になり、より局所的な解析が可能となり(Saffarian et al. 重要なことは、DNA分離法が微生物叢組成の評価に大きな影響を与えることである(Bajajら、2012;Hasanら、2016;Knudsenら、2016;Limら、2018;Nelsonら、2019;Thoendelら、2016;Wagner Mackenzieら、2015;Wesolowska-Andersenら、2014;Yuanら、2012)。したがって、便や組織について十分に開発され標準化されたプロトコールは、マイクロバイオーム研究のコンセンサスに貢献する。
しかしながら、固形組織サンプルのマイクロバイオーム組成の研究に課題がないわけではない。全組織分離株には大量の宿主DNAが含まれており、単細胞生物やウイルスの存在は影を潜めている。ポリメラーゼ連鎖反応(PCR)は少数塩基配列を同定するための貴重な手法であるが、マイクロバイオーム研究の分野では、望ましい手法としてショットガンメタゲノムシーケンスへと徐々に移行しつつある。ショットガンメタゲノムシーケンスでは、分離されたDNA中の全配列を解析できるため、より高い精度で種の検出が可能になる(Ranjan et al.) この手法のもう一つの大きな利点は、微生物種を識別し、潜在的な病原性因子を含む遺伝子含有量を解析できることである(Ranjan et al.) これは、病原体と常在菌を種レベルで識別するために極めて重要であると考えられる(Taddese et al.) 残念なことに、ヒト組織のマイクロバイオーム研究にショットガンメタゲノムシーケンスを適用することは、これらのサンプルに存在するヒトDNAの量が多く、細菌DNAよりも圧倒的に多いため、厳しく制限されている。
細菌とヒトのDNAの比率を改善するために、様々な方法が開発されてきた。これらの方法には、サイズによるヒト細胞の濾過(Marotz et al.、2018)、非メチル化CpGジヌクレオチドモチーフを標的としたヒトDNAの抗体媒介濾過(Horz et al、 2018)、ヒト特異的細胞溶解に続くDNA分解(Horz et al., 2010; Marotz et al., 2018; Nelson et al., 2019; Thoendel et al., 2016)のうち、後者が最も効率的な細菌DNA濃縮をもたらす(Marotz et al.) 細菌DNA濃縮は、少数種の同定とヒト組織サンプルに存在する微生物ゲノムの高いシーケンスカバレッジに貢献し、したがってこれらのサンプル中のマイクロバイオームの分類学的および機能的解析を改善する。
細菌DNA濃縮の注意点の1つは、DNA分離方法がマイクロバイオームプロファイルに影響を及ぼすことである(Biesbroekら、2012;Bjerreら、2019;Horzら、2010;Knudsenら、2016;Marotzら、2018;Nelsonら、2019;Thoendelら、2016)。細菌は溶菌に対する感受性が異なり、その結果、単離法中に早すぎる時期に溶菌する傾向がある細菌もあれば(Biesbroekら、2012;Horzら、2010)、ビーズビートによる機械的溶菌など、DNAを放出するために余分な工程を必要とする細菌もある(Limら、2018;Yuら、2017)。機械的溶解の追加は、グラム陰性菌の分離を損なうことなく、グラム陽性菌の分離を改善した(Biesbroekら、2012;Knudsenら、2016;Yuanら、2012)。さらに、ミュータノライシンを用いた酵素溶解は、グラム陽性菌を濃縮する可能性がある(Moen et al.) これらの戦略の最終的な目標は、細菌対ヒトのDNA比率を高め、サンプルの細菌組成を忠実に反映したDNA分離物を得ることである。
ヒトの腸内細菌叢の理解が飛躍的に進んだのは、その大部分が組織ではなく便サンプルに基づいているからである。そのため、宿主に最も近い場所に存在する細菌の研究は、腸内の局在性(例えば、結腸セグメントや腫瘍への局在性)に関する重要な情報とともに、ほとんど無視されてきた。ショットガンメタゲノムシーケンスに適した腸組織サンプルからの細菌DNA取得における現在の限界に対処するため、ここでは最適化されたDNA分離法を紹介する。我々の方法は、HMPプロジェクト(Gevers et al., 2012)を改良したもので、現在最も優れたDNA分離法である細菌DNA濃縮、ムタノライシン処理、熱ショック、ビーズビート法の重要な要素を組み合わせたものである。我々のプロトコールは、グラム陰性菌由来のDNAを維持しながら、グラム陽性菌を効率的に溶解する。われわれの最適化されたプロトコールは、~2~5mmの範囲の生検の細菌含有量を濃縮し、ショットガンメタゲノミクスを用いてin vivoで取得した組織生検に関する2つの大規模な前向き研究で検証された。この方法は、細菌マイクロバイオームの構成と機能の分野における再現性のある研究に貢献し、腸関連組織だけでなく、細菌が十分に発現していない組織にも価値がある。
2 方法
2.1 ヒト結腸生検の収集
2017年から2018年にかけて、ナイメーヘンのRadboudumc病理学教室で、オランダの法律に従って生体外残存切除結腸材料を入手した。切除した2つの結腸から約2mmの鉗子生検20個(患者1と2の生検10個)、切除した5つの結腸から約5mmの生検24個(それぞれ患者3~7の生検4個、2個、8個、4個、6個)を採取した。研究目的での冗長組織の匿名使用は、Radboudumcにおける患者との標準治療契約の一部であり、患者はこれを拒否することができるため、残存結腸切除の研究に研究倫理委員会の承認は必要なかった。切除された結腸は手術室から病理室に運ばれ、組織はサンプルを採取する前にdH2Oで洗浄された。対象患者の誰一人として残余材料の使用に対する異議申し立てはなく、すべての材料は匿名で処理された。生検は清潔なメスで切除され、推定5mmまでの生検が得られた。あるいは、生検鉗子を用いて約2mmの生検を行い、大腸内視鏡検査で採取した生検の代用とした。採取後、生検片は液体窒素中で凍結チューブに入れてスナップ凍結し、-80℃で保存した。
ショットガンメタゲノム配列決定のために生体内で採取した鉗子生検は、スクリーニングのための大腸内視鏡検査を受けに来た患者から採取し、2つの臨床前向き研究、すなわち、遺伝学的に確認されたリンチ症候群患者のみを対象としたBBC研究(NL57875.091.16)、または潰瘍性大腸炎患者と既知の大腸疾患のない患者を対象としたBaCo研究(NL55930.091.16)のいずれかに参加した。上行結腸(VR1)および下行結腸(VR2)において滅菌鉗子を用いて健康な外観の組織生検を2回採取し、オプションとして前癌病変または炎症が疑われる部位またはその近傍(VR3)において追加生検を1回採取し、直ちに滅菌チューブに入れて液体窒素中で採取した。すべてのサンプルは2017年から2018年にかけてRadboudumc Nijmegenで採取された。両試験は、内国歳入庁CMO-Arnhem Nijmegen(CMO 2016-2616およびCMO 2016-2818)およびRadboudumcの理事会により承認された。大腸内視鏡検査前3ヵ月以内に抗生物質を服用した患者は除外した。すべての患者は18歳以上で、インフォームドコンセントに署名した。生検は採取後直ちに液体窒素中で凍結管に入れ、-80℃で保存した。各分析に使用した研究ステップ、患者、生検の概要については、付録1の表A1を参照のこと。
2.2 細菌DNA分離プロトコール
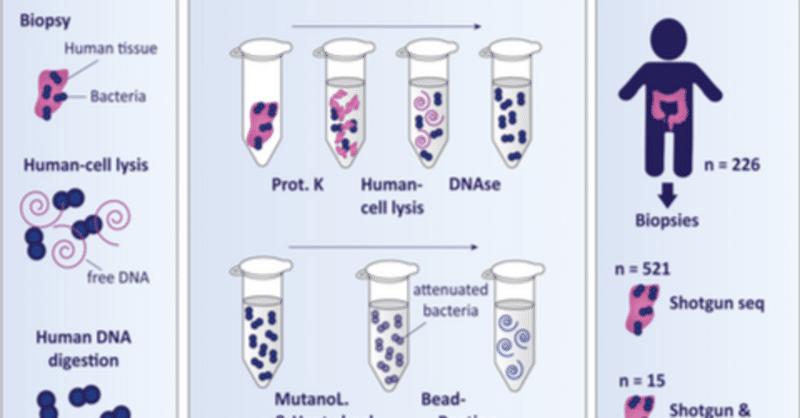
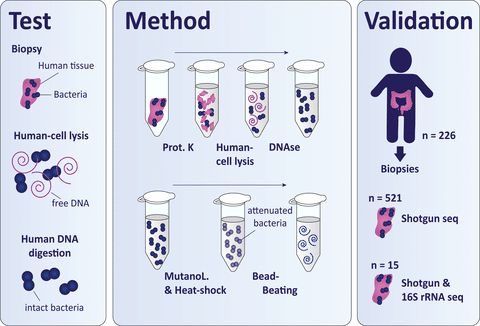
細菌DNA単離戦略では、ヒト細胞溶解およびDNAse処理(図1、上段)による細菌DNA濃縮を行い、それに続いて、以前に最適化したビーズビーティングプロトコル(図1、下段)を実施した(Couto Furtado Albuquerque et al.) ビーズビーティングプロトコルは本論文を通して変更しなかったが、細菌DNA濃縮のために2つの代替戦略を試験した。最初の戦略では、Molzym DNA分離(Ultra-Deep Microbiome prep, Molzym, 2020)キットを使用した。molDNAse不活性化ステップを含め、メーカーのプロトコールに従った。その後、qPCRでより高い細菌シグナルが得られることが示されたため、機械的な細菌細胞溶解を補助するためにビーズビーティングプロトコルを適用した(付録2の図A1)。第二の戦略として、タンパク質消化にプロテイナーゼK(19133、Qiagen)、宿主細胞の選択的溶解にサポニン(47036-50G、Sigma-Aldrich)またはトリトン(9002-93-1、Sigma-Aldrich)を含むリン酸緩衝生理食塩水(PBS)(Braun、220/12257974/1110)、宿主DNA除去にTurboDNAse(AM2239、Qiagen)を含む代替プロトコルを確立した。ヒト細胞の溶解について、異なる濃度の洗浄剤、トリトンまたはサポニンの効果を評価し、DNA分離工程に生検洗浄液(A点またはB点)を含める最適なタイミングを実験した(図1)。
詳細は画像に続くキャプションにある
図1
図ビューアで開く
パワーポイント
キャプション
細菌細胞の溶解には、0.5 KU/mL mutanolysin(SAE0092、Sigma-Aldrich)、ヒートショック、およびQiagen社のDNAeasy PowerLyzer PowerSoilキット(以前はMoBio社のMoBio PowerLyzer PowerSoil DNA単離キットとして知られていた)のバッファーC1での処理が含まれた。Magnalyser(Roche社製)を用い、6400rpmで20秒間ビーズビーティングを2回行い、その間に氷上で30秒間ビーズビーティングを行った。細菌溶解後、DNA分離キットのマニュアルに従った。最終的なプロトコルは付録3に示す。我々の最終的な細菌濃縮プロトコール(図1、ルートBおよび付録3)は、独立した研究室(ラドバウド大学水・湿地研究所)でもゼブラフィッシュの鰓から細菌を分離するために試験されたが、MoBio DNA分離キットの代わりにCTAB抽出を併用した(付録4)。
2.3 細菌の培養
Collinsella intestinalis (DSM13280)、Bacteroides vulgatus (3775 SL(B)10)、Escherichia coli (NTB5)、Streptococcus gallolyticus subsp. gallolyticus (UCN34)を、酵母エキスL-システイン・ビタミンK・ヘミンを添加したBrain Heart Infusion寒天培地 (BHI-S; ATCC medium 1293)で培養した。C. intestinalisとB. vulgatusは、嫌気条件下で48時間培養した後、液体培地に移し、37℃で48~72時間培養した。大腸菌とS. gallolyticusは、好気条件下でプレート上で一晩増殖させた後、BHIの液体培地に移し、37℃/5% CO2で24時間培養した。4600rpm、10分間の遠心分離で細菌をペレット化し、-20℃で凍結した。細菌ペレットを解凍し、光学濃度(OD at 620 nm)が1になるまでPBSに溶解し、そのうちの50 µlをTritonおよびサポニン処理による細菌のDNA放出を測定する実験に使用した。
模擬群集を作成するため、1 ODの細菌PBS懸濁液を400 µl(40%B.vulgatus、30%大腸菌、20%S.gallolyticus、10%C.intestinalis)に混合し、各実験条件ごとにペレット化した。
2.4 トリトンおよびサポニン処理による細菌のDNA遊離
細菌をPBSに溶解し、最終濃度0.1%、0.025%、0.0125%、0.006%の洗浄剤Triton(%v/v)またはサポニン(%w/v)を添加した。細菌は、洗剤またはPBSのみで、37℃で30分間インキュベートした。サンプルを10,000×gで10分間遠心し、Qubit dsDNA HS assay kit (Q32856, Thermo Fisher)を用いて、Qubit Fluorometer 2.0 (Thermo Fisher Scientific)でDNA濃度を測定した。Mann-Whitney U 検定を用いて、洗浄剤に暴露したサンプルと PBS に暴露したサンプルの上清中の DNA を比較した。
2.5 ヒト組織溶解に対するサポニン0.0125%の効果
サポニン0.0125%がヒト細胞の溶解を誘導できるかどうかを調べるために、推定サイズ5mmの切除ヒト結腸生検を、サポニンを用いた選択的細胞溶解のステップまで、我々の最適化したプロトコールに従って処理した(図1および付録3)。この最後のステップでは、細胞ペレットを0.0125%サポニンまたはターボDNAseバッファー中のPBSとインキュベートしたが、ターボDNAse酵素は使用しなかった。サンプルを37℃で30分間インキュベートして細胞を溶解し、上清を10,000×g、4℃で10分間の遠心分離を2回繰り返して細胞残渣を除去した。上清中のDNAを100%エタノールで沈殿させ、10,000×g、4℃で20分間遠心した。沈殿したDNAを70%エタノールで洗浄し、10,000×g、4℃で20分間遠心した。最後にDNAを風乾し、dH2Oに懸濁した。
2.6 16S rRNA の定量的リアルタイム PCR
各反応は、0.4 µM フォワードプライマー、0.4 µM リバースプライマー、1X Power SYBR Green (A4368702, Applied biosystems)から構成される。各反応のDNA量は、生検片の大きさが~5 mmの場合は1 ng、~2 mmの場合は0.1 ngであった。宿主(ヒトまたはゼブラフィッシュ)および細菌(すべての細菌、ファーミキューテス、バクテロイデーテス、γ-プロテオバクテリア、およびアクチノバクテリア)用のプライマーは、以前に使用され評価された(Albuquerque et al、 Amann et al., 1990; Bacchetti De Gregoris et al., 2011; Silva et al., 2009; Gorissen et al., 2009; Juretschko et al., 1998; Yang et al., 2015)。qPCRは7500 Fast Real-Time PCR system(Applied Biosystems®)を用いて行った。サンプルは50℃で2分間、95℃で10分間、95℃で15秒間、60℃で1分間のサイクルを30回繰り返した後、95℃で15秒間、60℃で1分間、95℃で30秒間、60℃で15秒間の連続サイクルを行った。すべてのqPCR解析は二重に行った。
モックコミュニティ(上述)から単離したDNAを陽性対照として用いた。付録 2 の図 A1 でのみ、ヒトの糞便から単離したリファレンス DNA を相対存在量のキャリブレーターサンプルとして使用した。ヒト血液から単離したリファレンスDNAは、バックグラウンドqPCRシグナルを設定するためのネガティブコントロールとして使用した。
2.7 qPCRの統計解析
サンプル間の細菌量の差を評価するため、サンプルのユニバーサル 16S rRNA シグナルを、陽性対照(ΔCt)(模擬コミュニティ分離株)のユニバーサル 16S rRNA シグナルを用いて校正した。倍差は2-ΔCtで計算した。メタゲノム解析の結果、最も一般的な門は、ファーミキューテス属(39.8%)、バクテロイデーテス属(16.7%)、放線菌属(9.3%)、プロテオバクテリア属(16.4%)、Verrocumicrobia属(0.2%)、その他(17.5%)であった(図4c)。その後、ΔCtをコントロールサンプルのΔCtと比較した(ΔΔCt)。倍差は2-ΔΔCtで計算した。対のサンプルは対のt検定で解析した。不一致のサンプルの場合は、Mann-Whitney U 検定を用いて比較した。どの洗剤がPBSと最も類似した細菌組成をもたらしたかを評価するために、フリードマン検定を使用した。すべての統計検定は、Graphpad Prism version 5.0を用いて行った。
2.8 ヒトin vivo大腸生検のショットガンメタゲノムシークエンシング
付録 3 に記載されているように、DNeasy Powerlyzer Powersoil kit(Qiagen)を含む最適化されたプロトコルを用いて DNA を単離した。DNA濃度は前述の方法で測定した。226人の患者から分離された合計521のヒト結腸組織DNAが、塩基配列決定のために香港のNovogene Bioinformatics Technology Co. サンプルは低インプットのNEBnextライブラリー調製を用いて処理され、ペアエンドシーケンスはIllumina Novaseq 6000でインサートサイズ350bp、リード長150bpで行われた。1サンプルあたりFastQフォーマットで1.2GBの出力データが保証された。サンプルはDNA濃度(Qubit)、コンストラクト長を測定し、ライブラリー調製の品質チェックを行った。13サンプルはライブラリー調製に失敗したため配列決定されず、その結果、224患者の508メタゲノムが配列決定に成功した(補足データS1: https://doi.org/10.5281/zenodo.4678214)。
さらに、16S rRNAとメタゲノム配列決定の比較のために、BBCの研究から、DNA収量が最も高かった患者12人の生検サンプル15個の第2セットを選択した。これらのサンプルの平均濃度は5.9 ng/µlであった。5μlは16S rRNA増幅に、残りはメタゲノミクス・ライブラリー調製に使用された。サンプルは香港のNovogene Bioinformatics Technology Co. メタゲノミクスの塩基配列決定は上記のように行った。16S rRNA遺伝子のV3-V4領域は、プライマー341F(CCTAYGGGRBGCASCAG)と806R(GGACTACNNGGTATCTAAT)を用いて増幅した。全てのPCR反応はPhusion® High-Fidelity PCR Master Mix (New England Biolabs)を用いて行った。ライブラリーはNEBNext® UltraTM DNA Library Prep Kit for Illuminaで作製し、QubitおよびqPCRで定量した。シーケンシングはIllumina NovaSeq 6000プラットフォームで行い、250 bpペアエンド生リード(Q30 > 94.8%)を生成した(補足データS2: https://doi.org/10.5281/zenodo.4678214)。
2.9 バイオインフォマティクス解析
品質管理、トリミング、アダプターの除去は、FastQCバージョン0.11.9およびtrimomaticバージョン0.35を用いて行った。BBMapバージョン38.84を用い、ヒトゲノムのGRCh38バージョンでヒトリードをフィルタリングし、アセンブルデータセットを作成した。フィルターしたリードはmetaSPAdes version 3.13.1でアセンブルした。bwaバージョン0.7.17とsamtoolsバージョン1.9を使用して、すべてのリードを分類されたコンティグとヒトゲノムにマッピングし、カバレッジ統計量を推定した。図4c+dの解析では、2.0e04以上の細菌リードを持つサンプルのみを使用し、その結果、379/508(74.6%)のヒト大腸生検由来のメタゲノム(224人中203人に属する)が得られ、サンプルあたりの平均リード数は1,100万であった。このカットオフは、細菌リードからの信頼性の高いプロファイルの生成を保証するために使用された(Cattonaro et al.) このカットオフは人為的に決定されたため、全データセットで同じ解析を繰り返した(付録2の図A6a+b)。サンプルは、リード数が最も少ないサンプルに従ってリードを再サンプリングすることで希釈した。Shannon多様性(α)とUniFrac多様性(β)(Lozupone & Knight, 2005)は、属レベルのリードの分類学的分布から推定した。多様性指標と門レベルの分類は、大腸組織生検のシーケンスに基づいて選択された、シャノン多様性と門の存在量を報告する文献から得られた値と比較した。メタアナリシスは行わず、生データもダウンロードしなかったが、報告された指標をメタゲノム結果の比較対象として使用した。これらの基準を満たす研究は、16S rRNAアンプリコンベースのものであった(Djuric et al., 2019; Kiely et al., 2018; Momozawa et al., 2011; Watt et al., 2016)。さらに、15サンプルについて16S rRNAシーケンスとショットガンメタゲノミクスの直接比較を行った。ショットガンメタゲノミクスサンプルは上述のように処理した。16S rRNAシーケンスから生成されたペアエンドリードは、固有のバーコードに基づいてサンプルに割り当てられ、バーコードとプライマー配列を切断することによって切り詰められた。ペアエンドリードはFLASH(V1.2.7; Magoc & Salzberg, 2011)を用いてマージした。Qiime(V1.7.0)の品質管理プロセス(Caporaso et al., 2010)に従って、特定のフィルター条件下で生タグの品質フィルターを行い、高品質のクリーンタグを得た(Bokulich et al., 2013)。タグはUCHIMEアルゴリズムを用いて参照データベースと比較し、キメラ配列を検出した(Edgar et al., 2011)。すべての有効タグを用いて、Uparseソフトウェア(Edgar, 2013)で配列解析を行った。類似度97%以上の配列は同じOTUに割り当てられた。各代表配列について、MothurソフトウェアをSILVA Database (Wang et al., 2007)のSSUrRNAデータベースに対して実行し、各分類ランク(Threshold:0.8~1)での種のアノテーションを行った(Quast et al., 2013)。OTUの存在量情報は、最も少ない配列のサンプルに対応する標準化配列番号を用いて正規化した。その後、Shannon index 2.9とUniFrac distance 0.56の解析をこれらの正規化データに対して行い、ショットガンメタゲノミクスから得られたデータと比較した(補足データS2: https://doi.org/10.5281/zenodo.4678214)。
3 結果
3.1 組織マイクロバイオームの集団捕捉には、PBS洗浄を含む全組織消化が必要である。
細菌DNAを濃縮する市販のキット(Molzym, 2020)が入手可能であったため、まずこの方法をテストした。さらに、洗浄した組織(生検洗浄)からDNAを分離するだけで、微生物DNA分離物中のヒトDNAの大部分を回避できるという仮説があるため、生検洗浄のみで細菌分析に十分かどうかを検証した。これを検証するため、生検と生検洗浄を別々に、Ultra-Deep Microbiome prep kit(Molzym, 2020)を用いて、我々のビーズビーティングプロトコルと組み合わせて分離した。生検は、タンパク質消化、選択的溶解、ヒトDNAの除去を含む完全なプロトコルで分離した(Methods参照)のに対し、生検洗浄ではこれらのステップを省略した(図1、経路A)。生検洗浄と生検のDNA単離から、同様の普遍的細菌16S rRNAシグナルが得られた(付録2の図A2)。
興味深いことに、生検洗浄液には、一致した生検に残存する微生物叢と比較して、グラム陽性菌が比較的多く、グラム陰性菌が少ないようであったが、これは有意ではなかった(付録2の図A2)。そこで、完全なプロトコール(生検と同様)とプロトコールの一部(生検洗浄と同様、図1の経路A)を比較し、模擬群集に対する戦略1の効果を検証した。その結果、選択的細胞溶解とDNAse処理を含む完全なstrategy 1プロトコルは、不完全なプロトコルと比較して、γ-プロテオバクテリアのシグナルが平均15倍(p = 0.03)、バクテロイデーテスのシグナルが平均27倍(p = 0.03)低いことがわかった(付録2の図A3)。この結果は、モックコミュニティでしかテストされていないが、グラム陰性菌とグラム陽性菌の分離を不利にすることを示唆しているため、戦略1を継続することは容認できない。
3.2 サポニン0.0125%は宿主細胞の溶解には安全だが、細菌細胞の溶解には安全ではないようだ。
プロテイナーゼKによるタンパク質消化、洗剤による選択的ヒト細胞溶解、溶解後の宿主細胞DNAを除去するためのDNAse処理など、類似の、しかし微調整可能なステップを用いて、戦略2を確立した。まず、どの洗剤が細菌門の比率に影響を与えることなく、ヒト細胞を効果的に溶解できるかをテストした。そこで、異なる濃度のトリトンとサポニンで処理した場合、PBSと比較して、純粋培養の細菌DNA放出(eDNA)が生じ、組織生検の細菌門に影響を及ぼすかどうかを試験した。まず、Streptococcus gallolyticus (ファーミキューテス)、Bacteroides vulgatus (バクテロイデーテス)、Escherichia coli (γ-プロテオバクテリア)、Collinsella intestinalis (アクチノバクテリア)の純粋培養をトリトンとサポニンに暴露した(図2a)。C. intestinalisはすべての条件下で溶菌に抵抗性であったが、B. vulgatusとS. gallolyticusはTriton存在下で溶菌に感受性であり、濃度が高いほど多くのeDNAが得られた。Tritonは大腸菌とC. intestinalisのeDNA量には影響しなかった。サポニンは0.1%の濃度で大腸菌のeDNAを増加させただけで、マイルドな洗浄剤であることが示された。これらの実験から、サポニン濃度は0.025%以下、トリトン濃度は0.006%以下であれば、細菌の溶解に安全であることが示唆された。
詳細は画像に続くキャプションにある。
図2
図ビューアで開く
パワーポイント
キャプション
次に、トリトンとサポニンが2人の患者(患者1と患者2)から採取した20の一致した組織生検の細菌構成を系統レベルで変化させるかどうかを試験した。サポニン(0.0125%または0.025%)またはトリトン(0.025%または0.006%)を含むプロトコールを用いてDNAを単離し、ファーミキューテス、バクテロイデーテス、放線菌、γ-プロテオバクテリアの相対的な存在量を、洗剤なし(PBS)で行った単離と比較した。各動物門について、PBSとの距離が最も短い洗浄剤を1位とし、次いで2位、3位、4位とした(付録2の図A4)。サポニン0.0125%は、すべての細菌門でPBSとの存在量の差が最も小さかった(図2b)。トリトン0.006%とトリトン0.025%は有意に高い順位を示した(それぞれp < 0.05とp < 0.001)(図2b)。さらに、ファーミキューテス類とバクテロイデーテス類の比率は、サポニン0.0125%の条件でのみ維持された(付録2の図A5)。このように、サポニン0.0125%では、サンプル内の系統レベルでの相対的な細菌組成が維持され、宿主細胞の溶解に使用しても安全であると思われた。
第3に、サポニン0.0125%がヒト細胞の溶解を仲介するかどうかを、2セットの3組織ホモジネート(サイズ:~5mm、(図1)の生検プロテイナーゼK処理後のステップ)をPBSまたはサポニン0.0125%のいずれかに暴露することによって試験した。サポニンで処理した組織の上清には、PBSのみで処理した組織と比較して、2倍以上の量のeDNAが含まれていた(p = 0.05)(図2c)。これは、サポニン0.0125%に組織をさらすと宿主細胞が溶解することを示している。
3.3 戦略2はバクテリアからヒトへのシグナルを増加させる
ヒト組織のDNA放出後、放出されたDNAを分解するためにDNAse処理を行う必要がある。eDNAの分解により、上清中の遊離DNAは著しく減少した(図3b)。DNAse処理後のDNA収量の有意な低下は、qPCRにおける細菌シグナルの増加(p = 0.004)と関連しており(図3a)、これは組織DNA単離液中の細菌対ヒトDNA分画がより大きいことを示しており、細菌DNA濃縮を示唆している。
詳細は画像に続くキャプションに記載
図3
図ビューアで開く
パワーポイント
キャプション
次に、大腸内視鏡検査で採取された臨床生検(大きさ:〜2mm)を鉗子を用いて採取した切除大腸からの生検でプロトコルを検証した。2人の異なる患者から5組の生検を採取した。各生検ペアは同じ洗剤濃度で単離され、そのうち1つだけがDNAseで処理された。DNAse処理により、qPCRにおけるヒトのシグナルは0.53(CI:0.42-0.65)に減少したが、細菌のシグナルは6.8倍(CI:2.2-10.52)に増加した(図3c)。トリトン0.006%とサポニン0.0125%は、両患者で4倍以上の濃縮を与えた(図3c)。興味深いことに、洗浄剤を使用しない場合(PBSコントロール)でも、DNAse処理によって細菌のシグナルが濃縮された。これは、加熱と遠心分離を繰り返す間に起こりうるヒト細胞の溶解によるヒトeDNAの存在によって説明できる。最終的に、魚の鰓から細菌DNAを分離するために、独立した研究室で戦略2の細菌濃縮プロトコルを適用した。サポニン0.0125%およびDNAse処理の使用により、qPCRにおける細菌は2倍になり、宿主シグナルは135分の1に減少したことから、我々の濃縮プロトコルは再現性があり、より多様な組織に適用できることが示された(付録1の表A3)。
以上の結果から、0.0125%サポニンによる宿主細胞の溶解とDNAse処理を含む戦略2は、サンプル中のヒトDNAを減少させ、細菌シグナルを増加させることに成功していることが示された。
3.4 ショットガン・メタゲノミクスによるヒト大腸組織生検の細菌組成は、16S rRNA分析で以前に報告されたものと類似している
最後に、われわれの最適化した方法を、2つの前向き臨床研究(Supplementary Data S1: https://doi.org/10.5281/zenodo.4678214)において、in vivoで取得した大腸生検に適用した。細菌リードの範囲は0.24%-40.51%であったのに対し、ヒトリードは16.1-99.48%であった。解析の結果、細菌リードの数は、顕微鏡検査によって決定された細菌の存在量と有意に関連していた(KruskalResult, statistic = 38.310, p value = 4.8e-09)(図4a)。細菌量は、蛍光in situハイブリダイゼーション(ほとんどの細菌に対するプローブEUB338:5'cy3- GCTGCCTCCCGTAGGAGT-cy3'3)で染色したメタカルン固定パラフィン包埋ペア生検でスコア化し、2人または3人の独立した観察者が低、中、または高細菌量でスコア化した。細菌量スコアは、細菌対ヒトのリード比とも関連している(KruskalResult、統計量=37.278、p値=8.038e-09)(図4b)。
詳細は画像に続くキャプションに記載
図4
図ビューアーで開く
パワーポイント
キャプション
分類学的分類に十分なリードがあることを確認するため、少なくとも20,000の分類された細菌リードを持つサンプルを解析した(完全なデータセットの解析も付録2の図A6に示す)。メタゲノム解析の結果、最も一般的な門は、ファーミキューテス(39.8%)、バクテロイデーテス(16.7%)、放線菌(9.3%)、プロテオバクテリア(16.4%)、ヴェロクミクロビア(0.2%)、その他(17.5%)であった(図4c)。これまでのところ、組織サンプルからのマイクロバイオームのショットガンメタゲノミクスは、細菌のDNA収量不足が障害となっており、大腸生検のショットガンメタゲノミクスはこれまで報告されていなかった。ここでは、16S rRNAシーケンスで配列決定されたサンプルと我々のデータを比較した(表1)。その結果、細菌門の分布は同等であった。さらに、我々の研究のシャノン多様性(2.9)は、他の研究(2.4-3.7)の範囲内であった。最後に、我々の研究の結果、平均ペアワイズUniFrac距離は0.56であり(図4d)、これはMomoozawaらで報告されたUniFrac距離(0.55)と同様であった。
表1. 本研究(WGS)のヒト大腸生検のマイクロバイオームプロファイルは、これまでに発表されたものと類似している(16S rRNA)。
本研究 Djuricら Kielyら Wattら Momozawaら
記号 図4 青星 赤三角 赤十字 赤六角 赤四角
ファーミキューテス 39.8 61 52.5 46.5
バクテロイデーテス属 16.7 27.3 39 43.2
放線菌 9.3 2.2 - 0.5
プロテオバクテリア類 16.4 4.5 2.5 5.1 - - - - 0.5
疣贅菌 0.2 3.8
フソバクテリア 0.0 0.1 1.5 - - - - - - フソバクテリア
その他 17.5 1.1 4.5 4.7 - - シャノン指数
シャノン指数 2.9 3.5 2.4 3.7
I. シンプソン指数 5.0 20.3 - - 20
UniFrac d. 0.56 - - - 0.55
注
我々は我々のマイクロバイオーム・プロファイルを、Djuricら、Kielyら、Wattら、およびMomozawaらで報告されたものと比較した。これらの結果を図4c + dに記号で表した。また、シャノン指数、逆シンプソン指数(I. Simpson index)、およびUniFrac距離(UniFrac d.)が報告されている場合は、それを示す。
さらに、BBC研究参加者の追跡調査で得られた15個の追加生検は、16S rRNA配列決定とショットガンメタゲノミクスの両方で配列決定された。これらの15個の生検は、フォローアップの生検組織分離株のより大規模なプールの中で最もDNA収量が多かったため選択されたものであり、それによって同一サンプルの2つの配列決定法に対して十分な収量が得られた(補足データS2: https://doi.org/10.5281/zenodo.4678214)。門、綱、目、科、属のレベルでは、アンプリコンシークエンシングとショットガンは高い相関があった(ピアソン:r = 0.87, p = 1.80e-84)(図4eおよび付録2の図A7(綱から種のレベルまで))。種レベルでは低い相関しか見られなかった。シャノン多様性とUniFrac距離はシークエンシング技術間で有意差はなかった(図4f+gおよび補足データS2: https://doi.org/10.5281/zenodo.4678214)。注目すべきは、15サンプル中4サンプルがスピロヘータ症を示し、これが低いシャノン多様性指標の一因となっている可能性があることである。
細菌リードはまだ低いこともあるが、我々が最適化した細菌DNA分離プロトコール(戦略2)とショットガンメタゲノムシーケンスを組み合わせることで、これまでに報告された細菌組織プロファイルを再現することができ、両手法でシーケンスしたサンプルにおけるショットガンメタゲノムと16S rRNAシーケンスの直接比較では高い類似性が示された。我々の知る限り、大腸組織細菌プロファイルがショットガンメタゲノミクスで報告されたのはこれが初めてである。
4 結論
組織からの細菌DNA分離は、大量の宿主DNAのために複雑である。この問題に取り組むために、いくつかの戦略、プロトコール、市販キットが開発されてきたが、これまでのところ、組織細菌の解析に重要と考えられるすべての要素を考慮したものはなかった。本研究では、Molzym(2020)とHasanら(2016)、およびヒトマイクロバイオームプロジェクト(HMP)(Albuquerqueら、2017)にヒントを得て、0.0125%サポニンによる宿主DNAの選択的溶解とそれに続くDNAse処理によって細菌DNAを濃縮するプロトコルを開発した。その結果、培養細菌細胞の溶解を誘発することなく、また臨床生検サンプルの細菌組成を顕著に歪めることなく、4つの最も一般的な系統を代表する細菌DNA単離株が得られた。特筆すべきは、我々の戦略が魚のエラにも有効であることが示されたことで、他の組織にも同様に適用または調整できることである。
我々はまず、Ultra-Deep Microbiome prep kit (Molzym, 2020)とビーズビート法(戦略1)を組み合わせてテストした。ビーズビーティングを取り入れることで、すべての細菌門、特に放線菌の分離が促進された(付録2の図A1)。さらに、PBS洗浄を導入することでグラム陰性菌の検出が改善されることに気づいたが、これは本キットの細菌濃縮ステップ中にグラム陰性菌が早期に溶解することに起因すると考えられる(付録2の図A3)。この重要な限界は以前にも示唆されている(Loonen et al.)
我々が設定したプロトコル(ストラテジー2)は、糞便サンプルの処理用に開発したプロトコルの拡張版である(Albuquerque et al.) このプロトコールはHMPプロトコールから改変され、ムタノライシンによる酵素溶解ステップ、ヒートショック、ビーズビーティングを含む。我々のビーズビーティングプロセスは、糞便サンプルで最適化されている(Albuquerque et al.) 重要なことは、ビーズビートの速度と時間の微調整が、特定のビーズビーターごとに必要になる可能性があることである。ビーズビーティングが組織からの細菌DNA分離を改善するかどうかは疑問視されてきた(Carbonero et al. しかし、より最近の研究によると、ビーズビーティングはDNAの剪断を起こさず(Lim et al., 2018; Wagner Mackenzie et al., 2015)、組織分離株の余分な種の同定につながる(Yu et al., 2017)。我々のプロトコールや他の研究において、ビーズビーティングは、より高いDNA収率(Carboneroら、2011)、グラム陽性菌のより効率的な分離(Biesbroekら、2012;Knudsenら、2016)、細菌投入に最も近い群集構造(Yuanら、2012)、およびより高い微生物多様性(Limら、2018)をもたらすことが証明されている。これらの知見を総合すると、ビーズ・ビーティングを取り入れるべきであることが示唆される。しかし、ビーズ・ビーティングは、各研究室で最適化された適切な条件下で、適切な種類のビーズを用いて実施しなければならない。
我々のプロトコールにおけるもう一つの重要なステップは、分離株からのヒトDNAの除去である。これまでの研究では、唾液および歯肉縁下プラークサンプルでは、Molysis(Horz et al., 2010)を用いて約90%以上、鼻咽頭吸引液ではTurboDNAse(Hasan et al., 2016)を用いて90%以上のヒトDNA除去(qPCRによる)が報告されている。我々の結果は、組織生検におけるヒトDNA(qPCRによる)の約50%の減少を示した。TurboDNAseがうまく機能しているかどうかを検証するために、TurboDNAseがDNA分離物中のDNAを除去できるかどうかを試験した。その結果、TurboDNAseはDNA濃度を94%減少させた。われわれは、われわれのプロトコール中、大量のヒトDNAが依然としてDNAseを介した分解にアクセスできないことを結論づけた。興味深いことに、洗剤なしでTurboDNAseを使用すると、細菌対ヒトのDNA比も増加した。これは以前にも観察された(Hasan et al.) Hasanら(2016)の研究では、洗剤の使用により病原体と宿主のDNA比が高くなったが、我々の研究では洗剤の起因効果は明らかではなかった(図3c)。われわれの結果は、生検サイズの多様性、したがってヒトDNAの総量が影響していると考えられる。ヒトDNAシグナルが2倍減少すると、qPCRでは細菌DNAシグナルが7倍程度増加したことから、ヒトDNA量が細菌DNAシグナルを強く妨害していることがわかる。ヒトDNAが分離株中に残存していることは明らかであるが、我々はマイクロバイオーム・プロファイルの歪みを防ぐため、マイルドな洗浄剤(サポニン0.0125%)を選択した。
われわれのプロトコールは、われわれの研究目的(2つの前向き臨床研究における細菌マイクロバイオーム)には最適化されているが、他の研究目的には若干の修正が必要かもしれない。例えば、われわれのプロトコールの重要な部分は、細菌のDNAが細胞壁の分離によって保護されたままのDNAseステップであるため、このDNA分離プロトコールは、マイコプラズマのような細胞壁を持たない細菌を検出するには最適ではないかもしれない。このようなタイプの細菌を研究するには、別のアプローチが必要であるが、その中でも抗体を介した細菌DNAのろ過はまだ選択肢の一つであろう。プロトコールに少し手を加えるだけで、他の細菌の分離効率は落ちるものの、特定のサブタイプの細菌検出を改善できる可能性もある。例えば、連鎖球菌のDNA収量は、現在のプロトコールよりもさらに強力なビーズビーティングによってさらに高くなる可能性がある。注目すべきは、溶解剤としてサポニンを使用していることである。サポニンはコレステロールを標的にするため、DNAse処理の前に酵母の細胞溶解を誘導する可能性もある(Francis et al. このプロトコールの焦点は、マイクロバイオームの細菌成分の分離に設定されており、酵母に対してどの程度機能するかはテストしていない。従って、酵母を正確に表現するための改良が必要かもしれない。重要なことは、ショットガンメタゲノミクスシーケンスにより、すべてのサンプルから古細菌とウイルスが検出されたことである(補足データS1: https://doi.org/10.5281/zenodo.4678214)。
224名の患者508生検のショットガンメタゲノムシーケンスの結果、大腸組織の16S rRNAシーケンスデータに匹敵するシャノン多様性とUniFrac距離を持つ細菌プロファイルを作成できたことが示され、このシーケンス法が組織マイクロバイオームプロファイリングに使用できることが示された。とはいえ、我々の研究(ショットガン)と他の3つの研究(16S rRNA)の細菌組成にはわずかな違いが観察された。重要なことに、便サンプルのショットガンメタゲノミクスと16S rRNAを比較した別の研究でも、同様の違いが認められた。Ranjanらは、16S rRNAシーケンス(34%)よりもショットガンメタゲノミクス(14~21%)の方がBacteroidetesが少なく、16S rRNAシーケンス(0.4%)よりもショットガンメタゲノミクス(4~7%)の方がActinobacteriaが多いと報告している(Ranjanら、2016)。したがって、我々の研究と他の研究で観察された大腸組織マイクロバイオームの違いは、増幅バイアスに起因する可能性がある。
我々は成功したプロトコルの戦略を統合し、プロトコルに手作業で調整したステップを作成したが、ショットガンメタゲノミクスとアンプリコンシーケンスを並べて比較した場合の微生物プロファイルの保存性を確認するためには、さらなる試験が必要である。ショットガンメタゲノミクスと16S rRNAシーケンシングの両方を用いた15サンプルの比較では、種レベルを除くすべての分類学的レベルで、両手法間の細菌量の高い相関が示され、シャノン多様性とUniFrac距離も同等であった。濃縮ステップによってプロフィールが歪んでいないことを明確に結論付けるには、属や種レベルのより広範な分析が必要である。さらに、利用可能な材料が限られているため、いくつかの実験は小規模であり、模擬コミュニティは4つの異なる細菌種から構成されているだけであった。しかしながら、我々のプロトコールは、現在市販されているいくつかのキットよりも多くの知見を提供し、利用可能な研究に基づく16S rRNAシーケンシングと同等の結果で、組織ショットガンメタゲノミクスの応用を可能にする。
以上より、16S rRNA増幅工程を省略した大腸生検の組織ショットガンメタゲノミクスに使用できるプロトコルを初めて示す。我々のプロトコールは、グラム陰性菌の分離を維持するのに十分マイルドである一方、放線菌やファーミキューテス菌のような丈夫な細菌の分離を促進するステップも含んでいる。重要なことは、独立した研究室が魚の鰓に適用して実証したように、我々のプロトコールは他の組織から細菌を分離するために調整することもできるということである。言い換えれば、われわれのプロトコルは、便や結腸の組織サンプルの分析にすぐに使えるだけでなく、他の研究材料の分離プロトコルの基礎としても役立つ可能性がある。さらに、我々はショットガンメタゲノムシーケンスを選択したが、我々のプロトコルは16S rRNAアンプリコンシーケンスと組み合わせて使用することもできる。従って、我々のプロトコルは様々な研究環境に適用可能であり、広範な細菌の解析を容易にする。このようにして、我々のプロトコールは基礎的および臨床的なマイクロバイオーム研究に貢献し、健康と疾患におけるマイクロバイオームの役割をさらに明らかにすることができるであろう。
謝辞
本研究は、A. Boleijに授与されたオランダ科学研究機構(NWO)のVeni助成金016.166.089、およびC.E. Bruggeling、D.R. Garza、A. Boleijを支援したオランダがん協会(KWF Kankerbestrijding)の支援を受けた(KUN 2015-7739)。B.E. Dutilhは、オランダ科学研究機構(NWO)Vidi助成金864.14.004およびERC Consolidator助成金865694の支援を受けた: DiversiPHI。
利益相反
申告なし。
著者貢献
Carlijn E. Bruggeling: 概念化(分担)、形式分析(分担)、調査(分担)、方法論(分担)、原著(分担)、査読・編集(分担)。Daniel R. Garza: データキュレーション(代表); 形式分析(同等); 調査(同等); 方法論(代表); リソース(代表); ソフトウェア(代表); 検証(同等); 原稿執筆(同等); 査読・編集(同等)。スミア・アチュイティ 形式分析(支援)、調査(支援)。Wouter Mes: 形式分析(支援)、調査(支援)、検証(支援)、執筆-校閲-編集(支援)。Bas E. Dutilh: 概念化(均等)、資金獲得(均等)、方法論(均等)、資源(均等)、ソフトウェア(均等)、監督(均等)、執筆-校閲-編集(均等)。アンネマリー・ボレイ 概念化(lead); 形式分析(equal); 資金獲得(lead); 調査(equal); 方法論(equal); プロジェクト管理(lead); 監督(lead); 検証(equal); 可視化(equal); 原稿執筆(equal); 書評・編集(lead)。
倫理声明
本研究は、世界医師会倫理綱領(ヘルシンキ宣言)に従って実施された。臨床生検はBBC研究(NL57875.091.16)およびBaCo研究(NL55930.091.16)から取得した。両試験とも内国歳入庁CMO-Arnhem Nijmegen(CMO2016-2616およびCMO2016-2818)の承認を得た。参加者全員がインフォームド・コンセントに署名した。
付録1
表A1. 本研究における実験と材料の概略。各作用の簡単な説明を以下に示す。
プロセス アクション 材料 図
事前作業
ビーズビート(BB)とモルツィムの試験
Molzym +BBの試験 患者番号3の切除結腸の大きな生検(~5 mm)4枚 図A1(付録2) 2.
PBS洗浄液を用いたMolzym +BBの試験 結腸切除患者nr.4の大きな生検2枚 図A2(生検とPBS洗浄液を別々に分離したもの)(付録2) 3.
Molzym濃縮による細菌組成の試験 4つのモックコミュニティ 添付資料2の図A3
微調整可能なステップを持つ独立したプロトコールに変更する。
プロトコルのセットアップ
洗剤の選択
プロトコール条件下での細菌溶解試験 純粋細菌培養 図2a
どの洗剤がPBSと最も差が少ないかの試験 切除した結腸の患者番号1+2の鉗子(小)生検20例(患者あたり10例)a 図2b + Appendix 2の図A4 + A5
選択した洗浄液がヒト生検を溶解するかどうかの試験切除結腸の患者番号 7 の大きな生検 6 個 図 2c
プロトコールのセットアップ
細菌DNA濃縮の確認
PBS 洗浄を DNAse 処理に含めるべきかどうかの試験 結腸切除の患者番号 5 + 6 の大きな生検 12 個 図 3a+b
どの洗浄剤が最も強い細菌 DNA 濃縮をもたらすかを試験する 切除した結腸の患者番号 1 番と 2 番の合計 20 個の鉗子(小)生検(各患者につき 10 個)a 図 3c
バリデーション
我々の方法によるシーケンス結果
臨床in vivoで取得した224人の患者508人のヒト生検の画像化により、細菌リード数が細菌量を表しているかどうかを評価する 図4a+b
どの細菌群が存在するかを観察する
図4c+d
付録2の図A6
同じサンプルの16Sとショットガンメタゲノムシーケンスを比較する。
図4e、f、g
付録2の図A7
a 同じ材料と実験であるが、異なる側面が図に示されている。
表A1の説明:
本論文の目的は、ヒト組織から細菌DNAを分離するためのプロトコルを設定することであり、その際、以下の点に注意した:
すべての細菌(組織の近傍または内部に存在する)を含むように組織を完全に消化すること。
ヒトDNAを可能な限り除去する。
グラム陰性菌をプロセスの初期段階で溶解しすぎない。
丈夫なグラム陽性菌からDNAを取得するために必要なステップを含む。
16S rRNA増幅のバイアスを排除したシーケンスにより、再現性のある細菌プロファイルを作成します。
プロセスの説明(表中のアクション番号)
Molzym DNA分離キットが選択されたのは、ヒト組織からの細菌DNA濃縮法としてよく報告されているからである。私たちが個人的に開発したビーズビート(BB)プロトコルはHMPにヒントを得たもので、以前私たちの研究室で糞便用に最適化されたものである。我々は、主に放線菌のような丈夫なグラム陽性菌の分離を促進するために、BBを用いてMolzymをテストした。
PBS洗浄液(生検をボルテックスしたPBS)の細菌含量を、同じ洗浄生検の細菌含量と比較した。PBS洗浄液には前処理が施されておらず、グラム陰性菌がやや多く含まれているように見えたことから、Molzymの溶解バッファーがグラム陰性菌をプロトコールの初期段階で溶解しすぎているのではないかと考えられた。
モックコミュニティで前処理をした場合としない場合でMolzymをテストしたところ、やはり前処理によってグラム陰性菌が失われている疑いが浮上した。
そこで、溶解バッファーを独自に設計することにした。どの濃度のサポニンやトリトンが純粋な細菌培養に安全に使用できるかをテストした。
どの濃度のサポニンまたはトリトンが、切除生検における最も一般的なフィラの相対的存在量の変化を引き起こすかをテストした。
サポニン0.0125%が細胞溶解を引き起こすかどうかを、ヒトの切除材料をプロトコール条件にさらすことで検証した。
生検の洗浄をDNAse処理に含めるべきかどうかをテストした(洗浄によりヒト細胞が破壊され、上清中にヒトDNAが放出される可能性がある)。
どのような洗浄条件が最も優れた細菌DNA濃縮をもたらすかを検証した。
2件の前向き臨床研究のin vivoで取得したヒト生検についてショットガンメタゲノムシーケンスを実施し、プロトコルを検証した。細菌のリード数が、イメージングによって得られる細菌の存在量スコアと相関するかどうかを検証した。
大腸組織マイクロバイオームの一般的な細菌群(16S rRNAシーケンスで以前に文献報告されている)が、我々の方法で分離された(そしてショットガンメタゲノムシーケンスで処理された)サンプルにも存在するかどうかを評価した。
さらに15個の臨床生検(両方のシーケンス法を行うためにDNA収量が多い生検)で16S rRNAシーケンスとショットガンシーケンスを行い、細菌量、シャノン多様性、UniFrac距離を比較した。
表A2. qPCR用プライマー
ターゲット 順方向プライマー 逆方向プライマー 参考文献
ユニバーサルバクテリア 926F:AAACTCAAAKGAATTGACGG 1062R:CTCACRRCACGAGCTGAC Yangら(2015)&Bacchetti De Gregorisら(2011)
Firmicutes 928FirmF: TGAAACTYAAAGGAATTGACG 1040FirmR: ACCATGCACCACCTGTC Bacchetti De Gregoris et al.
バクテロイデス属 Bac960F: GTTTAATTCGATGATACGCGAG Bac1100R: TTAASCCGACACCTCACGG Yang et al.
γ-プロテオバクテリア
1080γF:
TCGTCAGCTCGTGTYGTGA
γ1202R: CGTAAGGGGCCATGATG Bacchetti De Gregoris et al.
放線菌
Act664
TGTAGCGTGAATGCGC
Act941R:AATTAAGCCACATGCTCCGCTヤンら(2015)
ヒトKRAS P696: AGGCCTGCTGAAAATGACTG P488: TGGATCATTCGTCCACAA Silva et al.
万能細菌(魚の鰓の実験に使用) 616F: AGAGTTTGATYMTGGCTCAG Eub338IR: GCTGCCTCCCGTAGAGT Juretschkoら(1998); Amannら(1990)
ゼブラフィッシュLepA遺伝子 GACTGCACACTGAAGGAATC Lep A gen: GCACTGTCCTCTAGAAAGC Gorissen et al.
表A3. サポニン0.0125%とTurboDNAseを用いた細菌濃縮により、qPCRにおける細菌/魚類DNA比が改善した。DNA単離は、DNAse処理を行った場合と行わなかった場合で行った。Ct値は上部に記載。下段には、DNA分離の有無によるシグナルの倍数差(FD)を示した。
濃縮あり(Ct) 濃縮なし(Ct)
バクテリアシグナル 宿主シグナル バクテリアシグナル 宿主シグナル
魚鰓分離株
32.08
35.47
35.94
29.13
27.95
30.45
31.02
31.58
28.25
30.17
33.01
33.22
23.47
22.96
平均 32.114 30.294 33.115 23.215
ΔCt=Ctあり-Ctなし
FD バクテリア(2-ΔCt) FD 宿主(2-ΔCt)
FD 2.001386775 0.0073962
1/fd 0.499653546 135.20456
付録2
詳細は画像に続くキャプションに記載
図A1
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A2
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A3
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A4
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A5
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A6
図ビューアで開く
パワーポイント
キャプション
詳細は画像に続くキャプションに記載
図A7
図ビューアで開く
パワーポイント
キャプション
付録3
プロトコール
細菌濃縮とビーズビートによる組織からの細菌DNA分離
参考文献 ヒト組織のマイクロバイオーム解析のためのDNA分離法の最適化 Carlijn Bruggeling1、Daniel R. Garza2、Soumia Achouiti1、Wouter Mes3、Bas E. Dutilh2,4、Annemarie Boleij1*。
目的
このプロトコールは、ヒト結腸組織サンプル(~2-5 mm)からの細菌DNA分離に最適化されている。細菌濃縮の際、生検をPBS中でボルテックスし、生検から細菌を放出させる。この上清(「生検洗浄液」)は、生検の残りをプロテイナーゼKを用いて細胞懸濁液にした後、サンプルに戻される。サンプルはヒト細胞を溶解する石鹸で処理され、外部DNAを消化するTurboDNAse処理と組み合わされる。その後、サンプル中の無傷の細菌をMutanolysinとヒートショックを用いて溶解する。最後に、ビーズビートを用いて機械的溶解を行い、それに続いて標準的なDNA単離操作を行う。
ここでは、段階的なプロトコルを提供する。
材料
PBS: Tris-HCL (220/12257974/1110, Braun).
Proteinase K(19133、Qiagen)。
サポニン0.0125%(47036-50G、Sigma-Aldrich)、PBS中、0.2 µmフィルター。
10×バッファー入りTurboDNAse(AM2239、Qiagen)。
Mutanolysin 10 KU、2 ml ddH2O中(SAE0092、Sigma-Aldrich)。
DNeasy PowerLyzer PowerSoilキット(Qiagen)。
(以前はMoBio Powerlyzer PowerSoil DNA分離キットとして知られていた)。
ビーズ溶液。
溶液C1~C6。
ビーズ(0.1 mm ガラスビーズ)。
2 mL コレクションチューブ 3 セット
スピンフィルター1セット
準備:
以下のことを確認する:
清潔な机(塩化物を含む)。
遠心機4℃。
70℃、37℃、65℃、95℃のインキュベーター。
氷バケツ。
ビーズビーター。
パート1:細菌の濃縮
PBS洗浄および宿主組織消化
1.5mlのエッペンドルフチューブを2セット用意し、うち1セットには500μlのPBSを入れる。
凍結バイオプシーを500µlのPBSで1.5mlチューブに入れる(ピペットチップを使用)。
チューブを5分間ボルテックスする(スピード8/9)。
PBS/プロテイナーゼKミックスを作る。
上清(「生検洗浄液」)を新しいチューブに移し、氷上で保存する。
生検が2mm以下の場合:197μlのPBSと3μlのProteinase Kを生検に加える。
より大きな生検の場合:180μlのPBSと20μlのProteinase Kを生検に加える。
短時間スピンダウンする。
サンプルを70℃、400rpmで15分間インキュベートする。
インキュベーターを37℃に設定する。
ボルテックスを短時間行い、組織がばらばらになるようにする。
700μlのPBSを「生検洗浄液」に加え、一致した生検(消化済み)に加える。
10,000×gで10分間スピンする。
Saponin/TurboDNAse/Bufferミックスを作る。
上清を捨て、ペレットを保存する。
宿主細胞の溶解とDNA消化
12. 生検1件につき100µlの混合液を加える:
88 µl サポニン
10 µl バッファー 10× Turbo DNAse バッファー
2 µl TurboDNAse(2単位/µl)
13. ボルテックスで15秒間懸濁する。
短時間スピンダウンする。
37℃で30分間400rpmでインキュベートする。
1.3mlのPBSを加える。
遠心(10,000×g、10分間、4℃)する。
ピペッティングにより上清を捨てる。
ムタノライシンミックスを作製する。
1 mLのPBSを加え、ボルテックスしてペレットを懸濁する。
遠心(10,000×g、10 分間、4℃)する。
ピペッティングにより上清を捨てる。
ペレットを-20℃で保存するか、ステップ23に進む。
パート2:ビーズビーティングのプロトコール
ビーズビートの準備
23. サンプルあたり180 µlのビーズ溶液+20 µlのミュータノライシンを加える。
ボルテックスで再懸濁する。
37℃で60分間400rpmでインキュベートする。
ヒーターを6℃に設定する。
チューブを400rpmでインキュベーターに入れる:
65℃で10分間、
95℃まで昇温(7分間)
95℃で10分間
室温まで冷却し、まもなくスピンダウンする。
ビーズビート:
28. 試料に550μlのPower bead溶液を加える。
チューブを30~40秒間ボルテックスする。
混合液をビーズチューブに加えます。
C1溶液(DNeasy分離キットの最初の溶液)60 µlを加える。
サンプルを冷やさないようにしますが、次のステップのために氷を用意します。
MagNA Lyserでビーズビートを行う:
6400 rpm、30秒
氷上で30秒間
6400 rpm、30秒間
サンプルを氷上に保つ
細菌DNA抽出
33. 10,000×g で 2 分間遠心する。
上清を新しい採血管に移す。
*最大総量は500 µlとする。
250μlの溶液C2を加え、5秒間ボルテックスし、氷上で5分間インキュベートする。
10,000×g で 1 分間遠心する。
600~800 µl を 2 ml 採血管に移す。
200μlのC3溶液を加え、軽くボルテックスした後、氷上で5分間静置する。
10,000×gで1分間遠心する。
上清750 µlを2 ml採血管に移す。
ペレット(~850 µl)を邪魔しないようにできるだけ多く加える。
溶液C4を振り、1.2 ml(2 × 600 µl)を加え、5秒間ボルテックスする。
できるだけ多く、~1 mlを添加する。
スピンフィルターに約675µlをロードし、10,000×gで1分間遠心分離する。
溶液 C5 を 500μl 加え、10,000×g で 30 秒間遠心する。
フロースルーを捨てる。
10,000×g で 1 分間遠心する。
新しい採血管にスピンフィルターを慎重に入れる。
50μlの溶液C6をメンブレンの中央に加える。
10,000×gで30秒間遠心する。
スピンフィルターを捨てる。
抽出したDNAを-80℃で保存する。
付録4
CTAB抽出
バッファー
100 mM Tris-HCl.
100 mM Na-EDTA。
1.5 M NaCl。
2% CTAB。
0.05 mg/mlプロテイナーゼK。
材料
10%SDS。
クロロホルム:イソアミルアルコール(24:1)
イソプロパノール
フェノール:クロロホルム:イソアミルアルコール(25:24:1)
3 M Na-acetate。
100% EtOH。
70% EtOH。
オートクレーブしたmilliQ H2O。
脱濃縮ゼブラフィッシュ鰓からのゲノムDNAのCTAB抽出
鰓サンプルをDNaseで消化した後、洗浄したペレットを100 µl CTAB抽出バッファーに懸濁し、37℃で30分間インキュベートする。
25 µl の10% SDSをサンプルに加え、よく混合し、65℃で1時間インキュベートする。5分ごとにチューブを反転させて混ぜる。
125μlのクロロホルム:イソアミルアルコールを加え、20秒間よく混ぜる。
サンプルを最高速度で15分間遠心する。
水相を清潔なチューブに移し、廃棄物はヒュームフード内の容器に捨てる。
0.6容量のイソプロパノールをサンプルに加え、-20℃で一晩インキュベートする。
サンプルを最高速度で15分間遠心する。
イソプロパノールを注意深く注ぎ落とす(ペレットを失わないように)。
500μlの70%EtOHでペレットを洗浄し、最大gで10分間遠心する。
エタノールを注意深く注ぐ。
チューブを5分間開けたままにして、残りのエタノールを蒸発させる。
ペレットを200µlのオートクレーブしたmilliQに懸濁する。
DNA抽出物のRNase処理
1 µl(10 mg/ml)のRNase Aをサンプルに加え、37℃で30分間インキュベートする。
200 µlのフェノール:クロロホルム:イソアミルアルコールを加え、20秒間完全に混合する。
最高速度で15分間遠心する。
水相を新しいチューブに移し、フェノール廃液をフュームフードの容器に捨てる。
2容量の100% EtOHと0.1容量のNaAcを加え、チューブを反転させて混合する。
20℃で1時間インキュベートする。
最高速度で20分間遠心してDNAをペレット化する。
500μlの70%EtOHでペレットを洗浄し、最高速度で10分間遠心する。
エタノールを注意深く注ぎ落とし、短時間の遠心で残りのエタノールをスピンダウンする。
ペレットを乱さないように、ピペッティングで残留エタノールを除去する。
エタノールがなくなるまでペレットを乾燥させる。
ペレットを50μlのオートクレーブしたmilliQ水に懸濁する。
PCR
qPCRプログラム
3:00 96°C 1×
0:15 96°C 40×
0:20 58°C
0:30 72°C
2:00 72°C 1×
qPCR ミックス
SYBR ミックス 2× 10 µl
フォワード (10 μM) 0.6 µl
リバース(10 μM) 0.6 µl
H2O ... µl (最大20 µl)
DNA 5 ng
オープンリサーチ
参考文献
文献の引用
PDFダウンロード
戻る
その他のリンク
ワイリーオンラインライブラリーについて
プライバシーポリシー
利用規約
クッキーについて
クッキーの管理
アクセシビリティ
ワイリーリサーチDE&Iステートメントと出版ポリシー
発展途上国へのアクセス
ヘルプ&サポート
お問い合わせ
トレーニングとサポート
DMCAと著作権侵害の報告
チャンス
購読エージェント
広告主・企業パートナー
ワイリーとつながる
ワイリーネットワーク
ワイリープレスルーム
著作権 © 1999-2023 John Wiley & Sons, Inc. すべての著作権はワイリーに帰属します。
この記事が気に入ったらサポートをしてみませんか?