
初心者の菌叢解析 Qiime2で解析(11)多様性解析 ~β多様性~

今回は多様性解析のもう一つ「β多様性」の解析を行っていきます。
多様性に関しましてはわかりやすい解説サイトが沢山ありますので、ご確認ください。
また、これまでの投稿につきましても合わせてご確認ください。
Qiime2のインストール方法から投稿しています。
α多様性解析につきましても興味のある方はご確認ください。
1.Sample depthの設定
β多様性を解析するにあたり、まずはSample depthの設定を行います。
設定したSample depthは各サンプルでどれくらいの菌種(Features)を確認したかに依存します。
以前作成した「table.qzv」ファイルで確認できます。
Qiime2 viewにtable.qzvをドラッグ&ドロップし、Feature countを確認します。
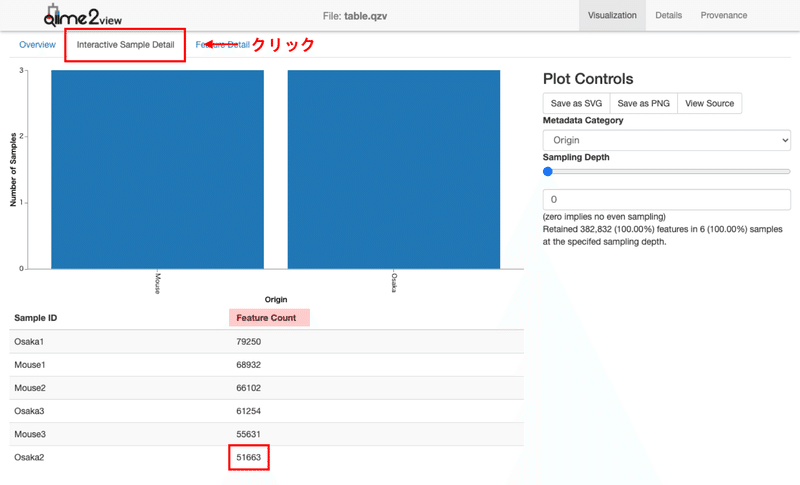
「table.qzv」ファイルの上の「Interactive Sample Detail」タブをクリックし、下のFeature Countの最小値を確認します。Osaka2の「51663」が最小の値になりますので、この値をSample depthに設定します。
シーケンスがうまく読めていない場合、この数字が極端に低いサンプルがあることもあります。その場合はそういったサンプルを除外することも考慮して、解析の方向性やSample depthを決める必要があると思います。
2.β多様性解析
次に実際にQiime2を動かしていきます。
まずは以下のコマンドで解析ファイルに移動します。
cd /Users/ユーザー名/Desktop/Qiime2_test
conda activate qiime2-2021.2以下のコマンドでβ多様性の解析を行います。
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 51663 \
--m-metadata-file sample-metadata.txt \
--output-dir core-metrics-results今回インプットしているqzaファイルは全て以前に作成したファイルになります。
「--p-sampling-depth」には先ほど確認したSample depthを入力します。こちらは解析毎に変化しますので、毎回確認してください。
解析が終了すると、「core-metrics-results」というディレクトリが生成されます。こちらの中に各解析結果が入っています。
qzvファイルはQiime2 viewで確認できるので、中を見てみてください。
「unweighted_unifrac_emperor.qzv」を確認してみます。

赤枠の中を選択することで、sample metadataで設定したグループ毎に色分けをすることができます。
それぞれの軸の中でサンプルがプロットされています。当たり前ですが、海洋の菌叢と腸内の菌叢ではその組成が全く異なることが分かります。マウスの腸内細菌叢のプロットは全て一箇所にまとまっています。
3.生データの出力
また、生データを取得して、自身でグラフを作成したい場合は以下のコマンドを入れてください。
qiime tools export \
--input-path core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--output-path core-metrics-results/unweighted_unifrac_pcoa_resultsこちらは生成されたqzaファイルやqzvファイルをエクスポートするコマンドで、解析したpcoaの結果をテキストファイルで出力します。
必要に応じて「--input-path」のファイルを変更したり、「--output-path」のファイル名を変更してください。
上記のコマンドで出力すると「core-metrics-results」ディレクトリの中に「unweighted_unifrac_pcoa_results」のディレクトリが生成され、「ordination.txt」というファイルが格納されていると思います。
「ordination.txt」をエクセルで開くと各主成分の数値が入っているので、その生データを元にグラフを作成してください。
平均をとって誤差範囲をつけることもできます。

4.有意差検定
次に得られた解析結果の有意差検定を行います。
ここでは「どのグループ間で有意差検定を行うか」という情報をコマンドに入れる必要があります。ここでいう「グループ」は「sample-metadata」で設定したグループ(metadata column)になります。
私の場合は「origin」というグループを「sample-metadata」に入れていますので、こちらで有意差検定を行います。
まずは「unweighted unifrac」で解析してみます。
以下のコマンドを入れてください。
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.txt \
--m-metadata-column Origin \
--o-visualization core-metrics-results/unweighted_unifrac_origin-significance.qzv \
--p-pairwise ポイントは「--m-metadata-column」の部分になります。
ここでは「Origin」と入っていますが、これは私の作成した「sample-metadata」の中で、今回解析したいグループになります。
語頭が大文字になっていますが、小文字にするとエラーが出ますので、注意してください。
Originというグループでの解析の為、最終的なファイルの名前も「unweighted_unifrac_origin-significance.qzv」にしています。必要に応じて各自で変更してください。
生成されたqzvファイルを確認してみます。

p値は「0.098」の為有意差は無いみたいです。
箱ひげ図の下にある(n=3)とかはサンプル数というよりは解析の計算回数のようです。(統計詳しくなくて申し訳ありません。)
エラーコード
以下のようなエラーコードが出た場合は「sample-metadata」を再確認し、「--m-metadata-file」のコマンドを入れ直してください。
今回は「Origin」を小文字の「origin」にした場合エラー出ました。
(1/1) Invalid value for '--m-metadata-file': There was an issue with
retrieving column 'origin' from the metadata.今回は以上になります。
この記事が気に入ったらサポートをしてみませんか?