
化合物を揃えて比べる

【ケモインフォマティクスのおやくそく】
塩はフリー体にしてからくらべましょう
化合物を比較する際、同じ化学構造の化合物は、基本的には同じ性質を示すと仮定します。
ただし速度論的溶解度、懸濁投与での経口吸収性など、同じ化学構造でもその化合物の状態(塩、溶媒和物、結晶化度等々)によって異なるプロファイルとなる場合もあるので注意は必要ですが。
化合物を比べる場合は、基本的には塩はフリー体にするなど構造正規化をします。
【構造正規化という課題について】
複数のソフトウェアによって生成されたバーチャルライブラリーや、外部の組織より入手した構造情報に関してとりわけ構造式表記の違いに起因する問題 が起こりがちです。また、ケミストによって塩の表記や短縮表記の使い方が異なる場合があります。
もともとは同じ化合物のデータが、多様な形態、あるいは表記法でデータベース化されているため、構造正規化の精度を高めようとすると果てしない闘いが始まります。
ケモインフォマティクスの重要な注意事項だと思うのですが、日本語のブログ記事は意外と見つけられなかったです。
そこで、構造正規化には具体的にどのような課題があるかと、その対策について公開されている情報をいくつか紹介してみようと思います。
【構造正規化ツールStandardizer】
創薬支援ITインフラとしてシステム化する場合、保守性も考慮して市販の専用プログラムを利用することが多いと考えます。
上で紹介したパトコア社のサイトから引用します。
Standardizerは標準化により化学構造式のあいまいさを排除します。
アロマタイゼーション/デアロマタイゼーション
水素原子表記の取扱い(表示/省略/立体を示す水素は省略しない)
メゾマーの変換(nitro,azide,etc)
トートマーの変換(oxo-enol,enamine-iminine,etc)
テンプレートに基づいたクリーニング
グルーピングの解除
塩、溶媒の除去(サイズ,リスト)
バッチ処理機能
ユーザー指定の標準化ルールのサポート
私の知る限り最も便利なツールです。でも有償ですから家庭用(?)ではないですね。
【玄人向け構造正規化技術】
無料で構造正規化をしたい方へは、やっぱりRDKitマスターの方が情報共有して下さっています。
例えば脱塩すなわちフリー体への変換についての記事の例です。
著名な玄人さんばっかりですね。
RDKit には Chem.SaltRemover という関数があるのですが、個人的にこれは使いにくいと思っています。
理由としては、
・前もって除きたい塩の種類を指定しておく必要があり、漏れが生じる可能性が高い。
・有機分子塩(マレイン酸塩など)が上手く除けない。
・入力の記法が独特でエラーを起こしやすい。
といった理由が挙げられます。
とより網羅的あるいは凝った前処理をしたい場合はPythonというかRDKitが有用です。
そして多分究極形の一つがGregにより提供されています。
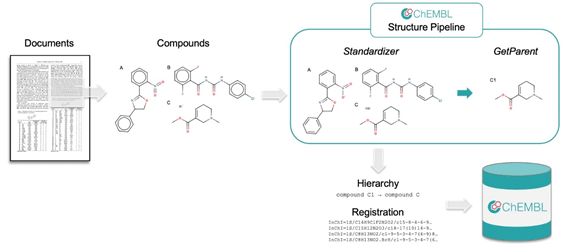
今やChEMBLに登録されるデータも本手法で構造正規化してもらえているそうです。
本当にただ畏敬の念をもって見上げています。
以下さらに余談です。ご興味あればご一読下さい。
【Beyond Rule of Five】
経口薬として好ましいかどうかを比べる際、分子量が重要な指標であることは確かです。
一方で、分子量のクライテリアとして500よりも大きくても経口吸収性のよい化合物は存在しています。
Beyond Rule of Five(bRo5)についてリピンスキー先生のコメントを引用しておきます。
今の創薬の最大の課題の中には、現在の設計空間を超えて考えなければならないものがあります。
ここでの主な問題は、多くの有望な薬物標的が小さい親油性リガンドを好まないということです。これは、多くの場合、結合部位が大きく、極性が高いか、機能がないため、従来のRo5リガンドは良好な親和力で結合しないためです。したがって、多くの医薬品開発者は、「ルールオブファイブを超えて」いる大きいリガンド(bRo5)を研究しています。
許容可能な経口薬物動態を達成するようにbRo5リガンドを促すことは驚くほど困難です。
そして政策研から興味深い報告が公開されています。
低分子医薬品の標的分子と分子量
-過去47年間の上市品からの調査-
「分子量500以上の上市品の標的分子のトレンド」まで解析されているのでご興味あればどうぞ。
ちなみに
抽出した低分子医薬品は塩構造を除いたフリー体構造として分子量を算出している。
と隅っこの方にちゃんと書いてあります。
【化合物をスコアリングするという研究分野】
TeachOpenCADDでは基礎技術を学ぶと言う意味でルールオブファイブを化合物フィルタリングに適用しました。
一方、今でもドラッグライクネスの指標は議論されています。
例えばですが、化合物を深層学習技術で多数発生させたとして、その化合物のスコアリング関数をどうしたら正確に定義できるのか。
創薬化学者は「AIに自分では思いつきもしないアイデアを出してほしい」と言うが、実際に提案された化合物を見ると「それは違う」と思うことが多いようです。
「では合成候補化合物の良し悪しをどう決めていますか」と聞かれても、数式化できるほど明確な定義は出来なくって、「じゃあどうやってアルゴリズムを改良すればいいんですか⁉」と膠着状態になったりしませんか?
今後も化合物をスコアリングするという旧くて新しい研究の進展に期待しています。
記事を読んでいただきありがとうございます。 先人の智慧をお借りしつつ、みなさんに役立つ情報が届けられたらと願っています。 もしサポートいただけるなら、そのお金はKNIMEの無料勉強会の開催資金に充てようと思います。