
定量PCR再考

今回は定量PCRについてもう少し詳しく説明していこうと思います。微量のDNAをPCRで限界まで増幅するのに何サイクル必要かといった話になります。コロナワクチンへのDNA混入疑惑についての関連記事とも言えます。専門的な内容になるのですが、宜しければお付き合いください。

https://anandamide.substack.com/p/sequencing-of-bivalent-moderna-and?utm_source=post-email-title&publication_id=456768&post_id=113965391&isFreemail=true&utm_medium=email
図1はMcKernan先生の定量PCRの対照実験です。未知サンプルの定量を行うために、段階希釈した既知量のDNAを標準サンプルとして用いています。
コロナ騒動により一般の方達にもPCRという言葉が広く知られるようになりましたが、コロナの検査で使われているのは定量PCRです。定量PCRはリアルタイムPCRとも呼ばれ、DNAの増幅の過程を蛍光シグナルによって同時進行で観測する実験技術です。二重鎖DNAへ結合し、緑の蛍光を発するSYBRグリーンを用いる方法や、増幅されるDNAに特異的なプローブを用いる蛍光プローブ法があります。McKernan先生は目的の配列のみを定量するために蛍光プローブ法を用いています。
定量PCRの利点は文字通り定量性ですが、定量PCRのPCR産物をクローニングや塩基配列決定等に利用するには不向きです。また、定量PCRは数kbの大きなPCR産物や複雑なPCRにも向いていません。そのため私が実験の現場でよく行うのは、定量PCRよりもむしろ通常のPCRです。目的や出発材料次第ですが、私は基本的にはPCRのサイクル数はできるだけ少ないように実験系をデザインします。
PCRの正式名称は「Polymerase Chain Reaction (ポリメラーゼ連鎖反応)」です。PCRは耐熱菌のDNAポリメラーゼを利用して、2つのプライマーで挟まれた特定のDNA領域を指数関数的に増幅する技術です。遺伝子を増幅する際にミスコピーが起きると突然変異となります。ちなみに、突然変異は英語でmutation (ミューテーション) で、遺伝子の変異といった意味です。突然変異と言っても日本語の「突然」のニュアンスは英語のmutationにはありません。そもそも突然変異は細胞内でも試験管内でも遺伝子を増幅する際につきもののトラブルです。例えば遺伝子をクローニングする際に、その遺伝子に突然変異が入っていると、大抵は実験を最初からやり直しになってしまいます。突然変異を最小限に留めるための工夫としては、変異率の低い酵素を使う、サイクル数を少なくして変異の起こる機会を減らす、などです。私自身の専門範囲として遺伝子クローニングは普段からよく行う実験ですので、どのくらいの量のDNAならばどの程度のサイクル数で十分量のPCR産物が得られるかについては、経験的に馴染みが深いです。
分子生物学の実験において、DNAを視覚的に確認するためによく使われるのが、アガロース電気泳動です。視覚化した際の画像では10 ng (ナノグラム) がギリギリ目視できる最小の量くらいでしょうか。100 ngくらいあればDNAをはっきりと確認できます。しかし10 µg (マイクログラム) 以上になるとかえって解像度は低くなり、確認しづらくなります。
µ (マイクロ) は10の-6乗。1 µgは0.000001 g。
n (ナノ) は10の-9乗。1 ngは0.000000001 g。
その下の単位はp (ピコ)、f (フェムト) です。
p (ピコ) は10の-12乗。1 pgは0.000000000001 g。
f (フェムト) は10の-15乗。1 fgは0.000000000000001 g。
クローニングや塩基配列決定などの分子生物学の実験に使われるDNAの量はngからµgくらいのレベルです。今は特別な顕微鏡を使えば1分子のDNAでも見る事が可能な時代ですが、現在でも普段の分子生物学の実験で使われるのはngからµgの程度の量なのです。ちなみにpg (ピコグラム) 量のDNAは定量PCRの対照実験くらいにしか私は使いません。
DNAの分子数を推定するためにまず実験で測定できるのはDNA濃度であり、そこから質量が求められます。しかし質量が同じであってもDNAの数は同じではありません。DNAのサイズが大きいほど質量当たりのコピー数は少なくなります。例えばゲノム遺伝子の場合1 µgが310000コピー、3000塩基対プラスミドDNAだと1 µgが3.2 x 10の11乗コピー、100塩基対のDNA断片だと1 µgが9.7 x 10の12乗コピーです。これを計算してくれるwebサイトもあります。
dsDNA: Mass to/from Moles Convertor
https://nebiocalculator.neb.com/#!/dsdnaamt
PCRで指数関数的にDNAが増える際、理想的に増えれば1サイクル当たり2倍になります。つまり、2サイクルで4倍 (= 2 x 2)、3サイクルで8倍 (=2 x 2 x 2)、5サイクルで2の5乗で32倍 (=2x2x2x2x2)、10サイクルで2の10乗で1024倍、20サイクルで2の20乗で約百万倍、30サイクルで2の30乗で約1億倍、40サイクルで2の40乗で約1000億倍です。
ただしDNAはPCRで無限に増え続けるわけではなく、ある量以上には増えなくなります。この状態を「プラトー」と呼びます。プラトーの原因はPCRの素材の枯渇やPCR酵素活性の低下などです。しかし、十分なDNA量からPCRを始めると、素材の枯渇前にプラトーに達します。PCR産物自体が新たなPCRを阻害するからです。二本鎖DNAが一本鎖に乖離した後にプライマーが鋳型に結合すると次のDNA複製反応が始まりますが、PCR産物が増えてくると、プライマーとの会合よりも一本鎖PCR産物がお互いに会合する割合が増えてきて、それが増幅反応を止めるのです。
経験的にはプラトーのDNA量は、50 µlのPCR反応液で1〜10 µg程度です。この値はPCR産物の大きさやプライマーなど条件によって変わりますが、およそµgのレベルです。
理想的な増幅効率の定量PCRの参考実験データを見つけたので紹介します。New England Biolab (NEB) の定量PCRキット、Luna® Universal qPCR Master Mixです。
NEB社は老舗の分子生物学の試薬メーカーです。制限酵素などの分子生物学の試薬が充実しており、タンパク精製やゲノム解析の製品までカバーしています。顧客への技術サポートがいつも的確で丁寧なので、私は大変信頼している会社です。

https://international.neb.com/products/m3004-luna-universal-probe-qpcr-master-mix#Product%20Information
図2の実験はNEBのqPCRキットの使用例です。ヒトcDNAからGAPDH遺伝子を増幅しています。未知のサンプルの定量を行うために、段階希釈した既知量のDNAを標準サンプルとして用いています。
https://international.neb.com/products/m3004-luna-universal-probe-qpcr-master-mix#Product%20Information
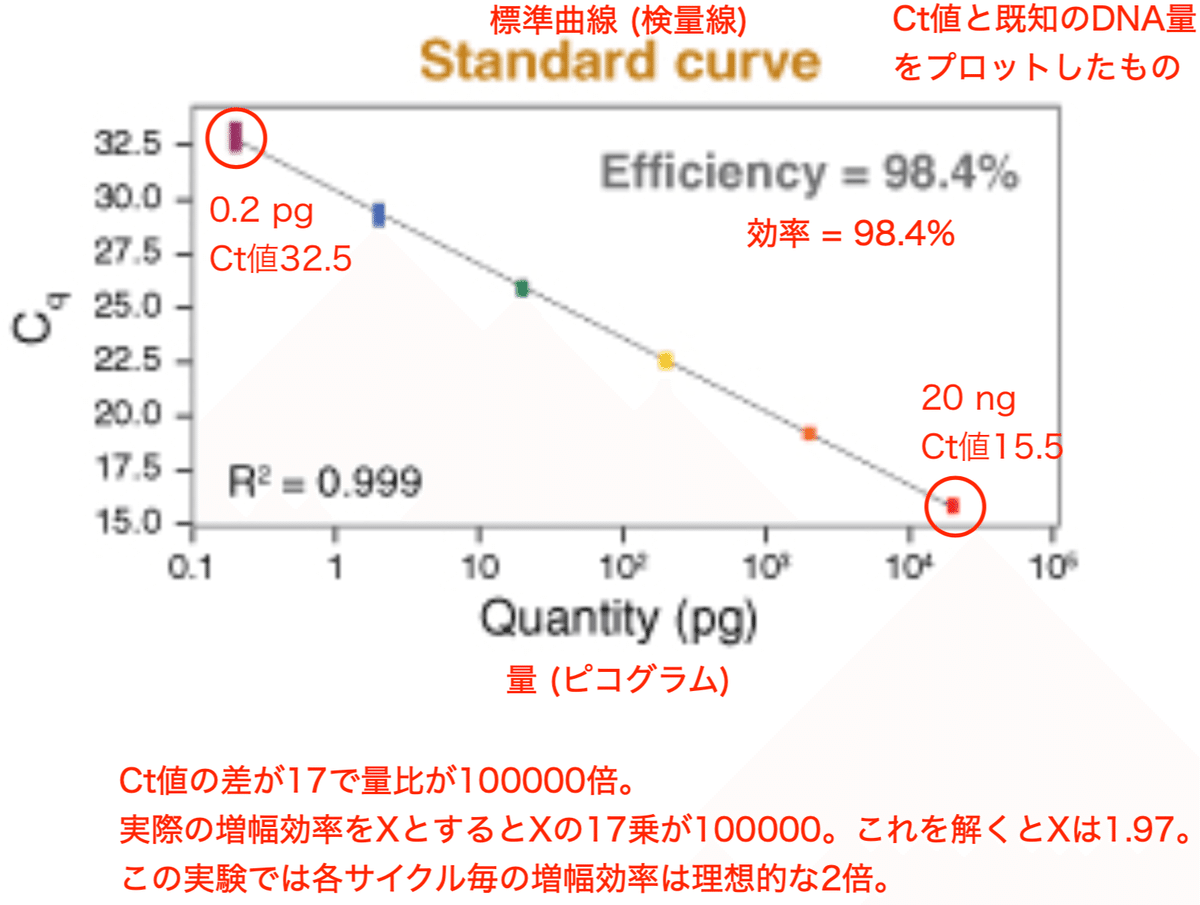
図3の検量線は図2のCt値とDNA量をプロットしたものです。検量線からサンプルのDNA量を測定できます。
この実験では検量線はほぼ直線になっています。20 ngのDNAだとCt値は15.5、0.2 pgのDNAだとCt値は32.5 (※2023年6月1日訂正、詳細はコメント欄参照))。Ct値の差が17で量比が100000倍。1サイクル当たりPCR増幅により理想的には2倍に増えるはずですが、実際にはPCRの効率は100%とは限りません。実際の増幅効率をXとして計算してみます。Xの17乗が100000とするとXは1.97。つまり、この実験では各サイクル毎の増幅効率は理想的な2倍となっています。それでも2〜20 ngのcDNAからの増幅で、PCR産物が最大量に達するには26サイクルが必要です。0.2 pgのcDNAだと42サイクル必要です。
図2の実験はほぼ理想的な条件で増幅がかかった実験ですが、多数の遺伝子が混ざったcDNAからのPCR増幅です。ではコピー数が既知のDNAを用いた定量PCRではどうでしょうか。

https://www.thermofisher.com/order/catalog/product/11733038

https://www.integra-biosciences.com/global/en/blog/article/qpcr-how-sybrr-green-and-taqmanr-real-time-pcr-assays-work

https://www.sigmaaldrich.com/IT/it/technical-documents/technical-article/genomics/qpcr/data-analysis
PCRの条件はプライマーや増幅領域の配列、PCR酵素に影響を受けます。図4〜6のように、25サイクル程度で最大量近くまで増幅するには10の6乗から10の8乗コピーの鋳型DNAが必要です。増幅効率はそれぞれの定量PCRによって変わってきますので、増幅効率を知るためには検量線での確認が必要です。必ずしも1サイクル当たり2倍に増えるわけではなく、実験系によってはより多い量のDNAが必要となります。

次に10の6乗から10の8乗コピーのDNAを質量に換算してみましょう (図7)。
一般的な定量PCRのアンプリコンのサイズが100~200 bpくらい、プラスミドベクターのサイズが3〜10 kbくらい、ファイザーコロナワクチンベクターのサイズが7.8 kbです。
理想的にDNAが増幅すると
20サイクル: 2の20乗 = 1,048,576倍 (約百万倍)
25サイクル: 2の25乗 = 33,554,432倍 (約3400万倍)
30サイクル: 2の30乗 = 1,073,741,824倍 (約10億倍)
35サイクル: 2の35乗 = 34,359,738,368倍 (約340億倍)
40サイクル: 2の40乗 = 1,099,511,627,776倍 (約1兆倍)
例えば、1 fgのDNAはプラトーまで増える頃には1 µg以上になっているのが通常ですので、10億倍以上の増幅が必要です。理想的な条件でも増幅には30サイクルかかりますし、実際にはプラトーの状態で増幅が鈍るので35サイクル近く必要です。実際には増幅は理想通りにいかない事も多く、必要なサイクル数がさらに多くなる事もよくあります。経験的には、相当量のDNAがなければ25サイクル程度ではプラトーには達しません。
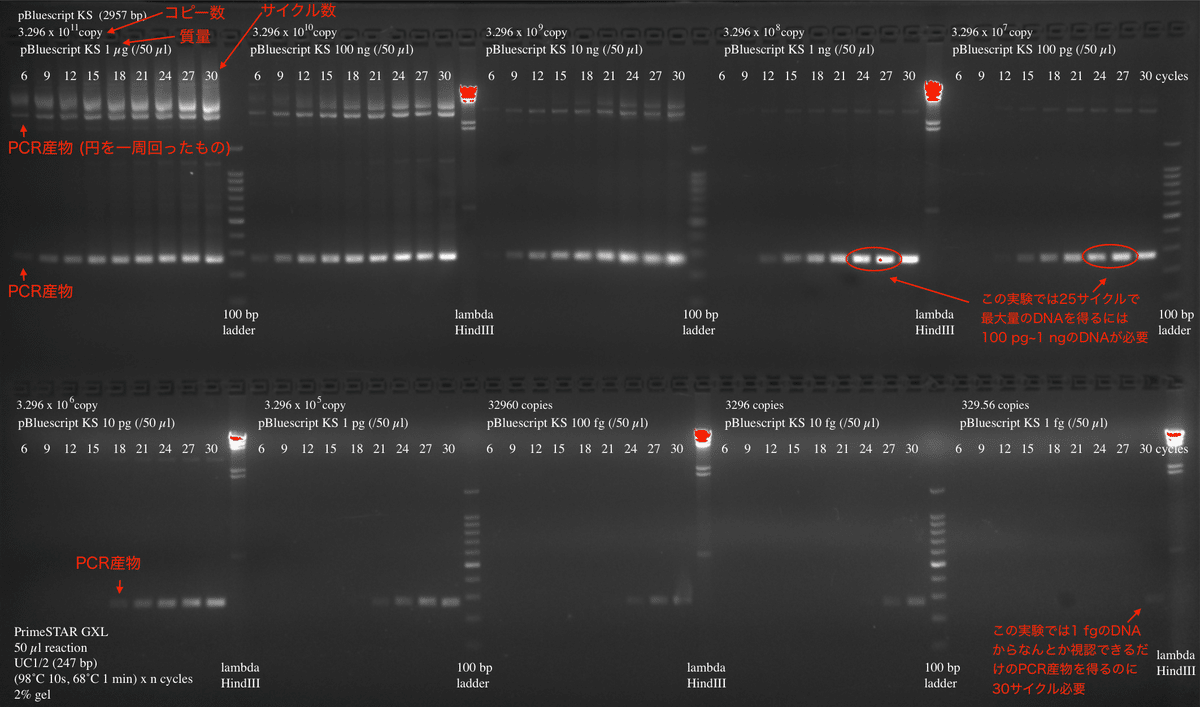
最後にもう1つの例として、通常のPCRの結果を示します。図8は私自身による半定量的PCRの実験です。これはコロナワクチンを使ったものではなく、標準的なプラスミドベクターを鋳型とした実験です。鋳型は約3 kbのプラスミドDNA (pBluescript KS)、50 µlの反応液中でのPCRです。PCRの結果は、定量PCRのようなグラフではなく、アガロース電気泳動により視覚化されています。
環状プラスミドからのPCRですので、円を一周したPCR産物も生じます。使用したDNA量とその分子数から、実際どれくらいの量のプラスミドDNAを出発材料にすれば、どのくらいのサイクル数でPCR産物が見えてくるのか、また、どれくらいのサイクル数で最大量に達するのかを実感していただければと思います。
この実験条件では、1 fgのプラスミドからは30サイクル後にようやくPCR産物が視認できるくらいです。25サイクル程度でPCR産物の量が最大に達するには、出発材料は100 pg〜1 ng必要でした。100 pg〜1 ngは1 fgの10万から100万倍です。25サイクルくらいで最大量まで増えるというのは、出発材料のDNAが「かなり多い」という事です。1 fg程度では通常は全く足りないのです。
一般論として、定量PCRのグラフに違和感を感じた場合などの参考になればと思い、今回このお話をさせていただきました。
この記事が気に入ったらサポートをしてみませんか?