クロストリジウム・ディフィシル(Clostridiodes difficile)を標的とする抗体ベースのエフェクター機構に関する知見を提供する工学的分泌性免疫グロブリンA

本文へスキップ
バイオレキシブ
ホーム投稿FAQブログアラート / RSSチャンネルについて
このキーワードで検索
検索
詳細検索
新しい結果 このプレプリントをフォローする
クロストリジウム・ディフィシル(Clostridiodes difficile)を標的とする抗体ベースのエフェクター機構に関する知見を提供する工学的分泌性免疫グロブリンA
https://pubmed.ncbi.nlm.nih.gov/37986930/

View ORCID ProfileSonya Kumar Bharathkar, View ORCID ProfileMichael J. Miller, View ORCID ProfileBeth M. Stadtmueller
doi: https://doi.org/10.1101/2023.11.08.566291
この論文はプレプリントであり、査読認証を受けていません。
00010011
概要全文情報/履歴メトリクスプレビューPDF
概要
分泌型(S)免疫グロビン(Ig)Aは優勢な粘膜抗体であり、クロストリジオイデス(Clostridioides difficile)を含む常在性微生物や病原性微生物と宿主との相互作用を媒介する。SIgAは高分子IgA構造をとり、分泌成分(SC)と結合している。SIgAは重要であるにもかかわらず、SIgAがどのようにして多様なエフェクター機構をサポートしているのかについては、ほとんど解明されておらず、SIgAを用いた治療法は存在しない。cSIgAは、C. difficile毒素に対する中和力の増加、細菌の凝集と細胞破裂の促進、細胞毒性の減少を示した。cSIgAはまた、C. difficileの形態学的変化と凝集現象を可視化および定量化することを可能にした。その結果、SIgAがC. difficile感染と闘うメカニズムが明らかになり、cSIgAの設計がこれらのメカニズムを調節できることが示され、幅広い抗原やエフェクター機構を標的とする可能性のある改変に対するcSIgAの適応性が示された。
はじめに
脊椎動物の上皮バリアは、その大部分が粘膜上皮によって形成されており、粘膜関連リンパ組織(MALT)[1]で組織化された特殊な粘膜免疫応答によって守られている。これらの応答は、一般的に遭遇する抗原に対する寛容と、病原体のクリアランスを促進するメカニズムとのバランスをとっているが、乱されると病原体の拡大、異常繁殖、疾病を引き起こす可能性がある[2]、[3]。粘膜抗体は、このバランスを維持する上で主要な役割を果たしている。ヒトでは、分泌型(S)免疫グロブリン(Ig)Aが優勢な粘膜抗体として機能しており、毎日腸内に分泌される抗体だけで数グラムを占めている[4]。
ヒトでは、IgAは単量体(m)、重合体(p)、分泌型(S)の形態で存在し、それぞれが異なるエフェクター機能を媒介する。mIgAは2本の重鎖(HC)と2本の軽鎖(LC)からなり、2つのフラグメント抗原結合(Fab)領域と1つのフラグメント結晶化可能(Fc)領域を形成する[5],[6]。mIgAは血清中で優勢であり、抗体依存性細胞貪食(ADCP)、抗体依存性細胞細胞傷害(ADCC)、抗体媒介性免疫調節[7],[8]など、Fc受容体(FcR)によって媒介される炎症性反応を開始することができる。一方、pIgAには2~5個のmIgAが含まれ、形質細胞は接合鎖(JC)と呼ばれるタンパク質でつなぎ合わせるが、ヒトにおけるpIgAの主流は二量体(d)である。すべてのpIgAは高分子イムノグロビンレセプター(pIgR)と結合することができ、このレセプターは複合体を上皮細胞の基底側表面から頂膜細胞表面へと移行させる。そこでpIgRエクトドメインはタンパク質分解的に切断され、pIgAと切断されたpIgRエクトドメインの複合体であるSIgA(分泌成分;SC)を粘膜分泌液中に放出する[9],[10]。
mIgAとは対照的に、SIgAのエフェクター機構は主に抗炎症性であり、病原体の除去と常在微生物に対する耐性のバランスをとっている[11]。この微妙なバランスは、十分に理解されていない生得的機序と適応的機序の組み合わせによって達成される[10]、[12]。適応的メカニズムは、微生物表面の抗原や、毒素などの細胞外病原性分泌物と親和性の高い相互作用を形成することができるFabsによって媒介される。抗原-SIgA複合体のサブセットは、宿主レセプターと相互作用する可能性があり、炎症反応に寄与する可能性がある[13],[14],[15]。SIgA相互作用の大部分は、微生物に対するSIgAのコーティングまたは架橋をもたらし、これは通常、微生物または毒素による上皮障壁の侵害を防ぎ、蠕動運動を介してそれらの除去を促進するメカニズムである免疫排除をもたらす[16]。分子レベルでは、SIgAは1つの微生物上の複数の抗原(例えば、表面タンパク質や炭水化物)の架橋、または2つの微生物上の同一の抗原の架橋を媒介することができる。微生物の架橋はSIgAの高分子構造によってサポートされると推定され、少なくとも2つの異なるメカニズム、古典的凝集、または連鎖成長によって起こりうる。古典的凝集は、SIgAでコートされた微生物同士の衝突によって生じ、微生物同士がくっつく。連鎖成長は、SIgAでコートされた母細胞が分裂し、その結果、娘細胞が結合したままになることから生じる。連鎖成長では、衝突ではなく連続的な分裂により、架橋した微生物の塊ができる。凝集と連鎖成長は相互に排他的ではなく、微生物の形態や濃度、抗原の発現や分布、さらには抗体の親和性や位置や濃度などの他の要因を含む多くの要因に依存する[16],[17]。SIgAの自然免疫様機構は、主にHC、LC、JC、SC、NおよびO結合型糖鎖のようなSIgAの構成部分との非特異的相互作用によって媒介されると考えられており、免疫排除にもつながる非特異的相互作用によって受動免疫を提供すると考えられる[12],[18],[19]。SIgAの生得的および適応的メカニズムは、共に微生物の宿主上皮細胞への付着を制限し、病原体の排除を促進し、あるいはある種の常在菌のコロニー形成を促進することができる。微生物へのSIgAの結合は、遺伝子発現、分裂、その他の生理学的プロセスを変化させる可能性があり、微生物に様々な影響を与えることができることも注目に値する[16]、[17]。これらのプロセスはすべて、SIgAのユニークな分子構造によって支えられている。SIgAは、Fabsの位置を拘束し、2つのFcR結合部位とSCを立体的にアクセス可能な状態に残す、複雑な構成要素の非対称配置によって特徴づけられる[17]。しかしながら、SIgAの構造と機能の関係はまだ十分に理解されていない。
SIgA-抗原相互作用の背後にある機序を解読することは、膨大な数の疾患を理解し治療する上で極めて重要であるが、エフェクターの機序や結果が多様であることから、そのためには多くの抗原特異的過程を理解することが必要である。特に興味深いのは、Clostridiodes difficile感染症(CDI)を引き起こすヒト腸管病原体Clostridiodes difficileとSIgAの相互作用である。CDIは、第一世界の国々で最も流行している院内感染の一つである[20]。典型的には、この疾患は上皮バリアを損傷して偽膜性大腸炎を引き起こし、その結果、初感染時のCDI関連死の2.7%、再発時のCDI関連死の25.4%をもたらす。さらに、CDI回復患者の35%が再発し、一度再発した患者の65%がさらに再発を繰り返す[21]。バリア障害は主にクロストリジウム・ディフィシルの毒素であるTcdAとTcdBによって引き起こされ、これらの毒素は主要な病原因子であり、宿主細胞に対して細胞毒性を示すが[22]、C. difficileの急速な増殖と、治療時の病原体の除去が不完全であることによって、この疾患は悪化する[20]。メトロニダゾール、フィダキソマイシン、バンコマイシンを含む標準的なCDI治療は、しばしば病原体を完全に除去することができず、常在菌種を標的とするため、腸内環境の異常が長期化し、再発を引き起こす;また、C. difficileの毒素であるTcdAとTcdB、およびC. difficileの芽胞を標的とすることができず[23],[24]、これらはいずれも再発の一因となる[25]。最近、TcdBを標的とするモノクローナル抗体(mAb)であるベズロトクスマブが、抗生物質治療とともに静脈内に投与された場合、TcdBの細胞毒性を制限できる最初のIgG1 mAbとしてFDAに承認された[26],[27]。しかしながら、ベズロトクスマブはC. difficileの増殖を抑えることができず、静脈内投与というアプローチ[26]に基づくと、腸管上皮細胞の先端側で毒素を中和したり排除したりするのではなく、おそらくすでにダメージが生じた後に、内在化した毒素を標的にしている可能性が高い。ヒトの宿主はC. difficile抗原を標的とするSIgAを発現しており[28]、SIgAがCDIに対する自然な防御を提供し、治療の可能性があることを示唆している。しかし、C. difficileのクリアランスを成功させたり、毒素の細胞毒性を制限したりするSIgA依存性のエフェクター機構は、まだ十分に解明されていない。
まとめると、SIgAは事実上、抗体マルチツールである。しかし、このツールの各部分が多様な粘膜抗原に対してどのように作用し、どのように健康に役立てることができるかについての知識はまだ限られている。ここでは、構造ベースの工学的手法を用いて、(1)導入された単一ドメイン抗体(sdAb)による抗原結合の増強、または(2)導入された単量体蛍光タンパク質(mFP)による検出の増強のいずれかをもたらす、新規な二機能性を有する一連のキメラSIgAを作製することにより、この課題に取り組む。我々は、天然およびキメラ(c)SCとSIgAのライブラリーを用いて、C. difficileとその毒素のSIgA依存的中和を調べ、cSCとcSIgAが中和を増強する可能性を評価した。その結果、SIgA依存的なエフェクター機能に関する機構的な洞察が得られ、SIgAが単量体よりも優れていることが示された。これらの知見は、多様な抗原に適用可能なSIgAエフェクター機構に新たな光を当て、疾患の調査および治療のためのcSIgAツールを開発する機会を提供するものである。
結果
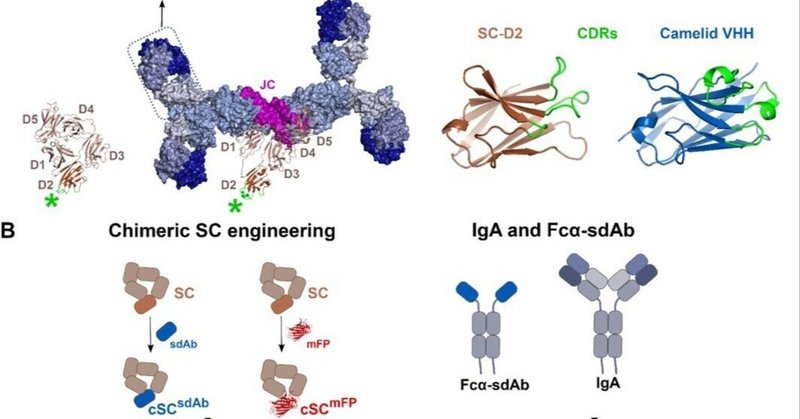
SCおよびSIgAの構造から、SC D2およびその相補性決定領域(CDR)様ループの大部分は、非配位型および配位型ともに溶媒にアクセス可能であり、pIgAとの結合には直接寄与しないことが明らかになった[29],[30],[31],[32]。この可能性を探るため、我々はD2をsdAbまたはmFPで置換した発現構築体を作製した(図1B)。これらのストラテジーを用いて、それぞれ固有の抗原と結合するsdAbを含む、あるいはmFPを含むキメラSC(cSC)のライブラリーを作製した。さらに、cSCをJCおよびIgA(またはFcα融合sdAb)変異体と共発現させ、キメラSIgA(cSIgA)のライブラリーを作製した。このライブラリーでは、cSCは抗原を認識するか蛍光を与え、Fab(または融合sdAb)は同じ抗原または異なる抗原を認識する。これらの戦略は、SIgAのコンフォメーションの利点(例えば、曲がり、傾き、活性)を維持しながら、付加的なモジュール機能を提供し、カスタマイズされた修飾に基づくcSIgAの標的に対する中和能と効果を研究するための多用途ツールとしてcSIgAを使用することを可能にした。
図1:
図をダウンロード
新しいタブで開く
図1:
キメラSCとSIgAの工学的戦略。
(A)SC、SIgA、ラクダ科IgG2の模式図、およびSC(PDB: 5D4K)とSIgA(PDB: 7JG2)の構造図。SCは5つのIg様ドメイン(D1-D5;茶色)を持ち、それらは閉じた非結合型コンフォメーションと開いたSIgA-結合型コンフォメーションで存在する。D2(暗褐色)のCDR様ループ(緑色)は、どちらのコンフォメーションでも溶媒にアクセス可能である。SIgAは1つのSC、1つのJC、そして2つのIgAモノマーから構成され、2対のFabsと2つのFcsを生じる(表示)。SC-D2(茶色)とラクダのVHH(青色)の構造およびそれらのCDR(緑色)を、CDRが同じ方向に向いていることを示す整列した構造とともに示す。(B)D2をsdAbsまたはmFPで置換したcSCを、JCおよびIgAまたはFcα融合sdAbと組み合わせて、cSIgA変異体を作製した模式図。
SCのD2はsdAbsで置き換えることができる。
CDIの病原性因子を標的とする目的で、我々はC. difficileの大型クロストリジウム毒素(LCT)、TcdAおよびTcdBを認識するsdAbを組み込んだ、あるいは他の抗原を認識するcSCおよびcSIgA変異体を作製した。得られたcSCのSEC溶出プロファイルが単分散であることから明らかなように、得られたcSCは広範囲のsdAb、VHおよびVLドメインに対して高い耐性を示した(図S1A)。標的抗原に結合するcSCの能力を調べるため、D2をsdAbに置き換えたcSC変異体、sdA20.1(cSCA20.1と呼ぶ)に着目した。sdA20.1はTcdA複合反復オリゴペプチド(CROP)ドメインと結合し(図2A)、推定CROPレセプター結合モチーフと宿主上皮細胞レセプター間の相互作用をブロックし、それによって毒素の内在化を防ぐと考えられている[33],[34]。
図2:
図をダウンロード
新しいタブで開く
図2:
cSCとcSIgAはTcdAと結合し、中和する。 A)TcdAドメイン構造の模式図(上)とTcdA-sdA20.1構造(下;PDBコード:7U1Zと4NBYの複合構造)。TcdAドメインは模式図と構造図において同じ色で、sdA20.1はマゼンタ色で示されている。(B) cSCA20.1、TcdAフラグメントTXA1、およびcSCA20.1-TXA1複合体の模式図と分析SEC溶出プロファイル。(C)cSA20.1Fcα、TXA1、およびcSA20.1Fcα-TXA1複合体の模式図とSEC溶出プロファイル。(D)mFcα-A20.1、TXA1、およびmFcα-A20.1-TXA1複合体の模式図とSEC溶出プロファイル。分析的SECは、等モル量のTXA1とcSCA20.1、cSA20.1FcαまたはmFcα-A20.1を室温で1時間インキュベートし、その後混合物を4度に冷やしてからSECを行うことによって実施した。同様に、TXA1、cSCA20.1、cSA20.1FcαおよびmFcα-A20.1も、比較のために個別にSECにかけた。(E)TcdA中和アッセイに用いた抗体の概略図。(cSCA20.1およびcSA20.1IgA-CDB1は、sdA20.1およびmFcα-A20.1の中和能力と比較したキメラである。(G)50pMのTcdAの濃度依存性中和。異なる数のsdA20.1部位を含む異なるキメラを用いて、TcdA中和のための結合部位数を増加させる可能性を検証した。図S1も参照。
具体的には、cSCA20.1(非リガンド)とcSA20.1Fcα(リガンド)を作製し、Fabsを欠くcSIgAを作製し、分析的SECを用いてTcdA-CROP断片であるTXA1との結合を試験した(図2Bおよび2C;図S1C)。その結果、TXA1-cSCA20.1複合体およびTXA1-cSA20.1Fcα複合体に対応するピークがコントロールよりも低い保持体積で検出され、cSCA20.1がTXA1抗原と非結合型および結合型の両方で結合できることが示された。sdA20.1と呼ばれる対応するsdAbとmFcα-A20.1と呼ばれる単量体Fcα融合変異体も、コントロールとしてAnalytical SECで検証されたin-vitro結合を示した(図2D; 図S1B-C)。これらの結果は、SC-D2をsdAbsで置換することが、SCおよびSIgAに抗原結合能を付加する効果的な手段であることを示している。
cSCおよびcSIgAは毒素を中和できる
cSCとcSFcαにsdAbとその結合特異性を付加する手段を開発した我々は、(1)非リガンドおよびリガンドバージョンのcSCA20.1、および(2)異なる数の抗原結合部位(sdAbsまたはFabsによって提供される)を含む天然およびキメラ変異体(例えば、cSCでは1部位、mIgAでは2部位、SIgAでは4部位以上、cSIgAでは5部位以上)の中和能を試験することを目的とした。SIgAおよびcSIgAの場合、抗原結合特異性はcSC、Fabs、またはFcαに結合したsdAbsによって提供される。Fabsは典型的な哺乳類の抗原結合部位を提供するが、sdAbsをFcαに融合させれば、キメラ変異体において同一の抗原結合部位の数(例えばsdA20.1の数)を増やすことが中和効力にどのように影響するかを調べることができると考えた。そこで、FcαとsdA20.1(cSCA20.1に含まれるsdAbと同じ)を融合したmFcα-A20.1を作製し、分析SECを用いてmFcα-A20.1とTXA1との結合を確認した(図2D;図S1C)。さらに、sdA20.1、cSCA20.1、mFcα-A20.1、分泌型-SFcα-A20.1、キメラ分泌型-cSA20.1Fcα-A20.1、およびキメラSIgAであるcSA20.1IgA CDB1を作製した(図2E)。cSA20.1IgA-CDB1に含まれるFab、CDB1-Fab[35]はTcdAに結合しないため、Fabが存在するリガンドコンフォメーションにおけるcSCA20.1の効力を試験する手段となる。
TcdAを標的とするキメラ抗体バリアントの中和能力を測定するために、Vero細胞を用いた細胞毒性中和アッセイを行った。このアッセイでは、Vero細胞培養を様々な濃度の抗体と一定濃度の50pM TcdAとともにインキュベートし、約65時間後の細胞生存率をアラマーブルーアッセイで測定した[36]。50%以上の細胞生存率は、毒素が中和されていることを示す。50pM TcdA単独では、抗体非存在下でVero細胞を約100%死滅させる。
まず、cSCA20.1(非結合cSC)、cSA20.1IgA-CDB1(結合cSC)、mFcα-A20.1、sdA20.1、およびSC(非結合コントロール)を比較する細胞毒性中和アッセイを行った。中和曲線から、cSCA20.1とcSA20.1IgA-CDB1はTcdAの細胞毒性作用を容易に中和できるのに対し、野生型SCは中和できないことが示された(図2F)。しかし、sdA20.1単体では、毒素断片の結合が認められるにもかかわらず、TcdAを中和することができなかった(図2F)[34]。注目すべきは、cSCA20.1(A20.1の1コピー)を含むキメラはmFcα-A20.1(A20.1の2コピー)と同様に機能したが、sdA20.1単独では中和できなかったことである。このことは、cSCA20.1、cSA20.1Fcα、mFcα-A20.1のような立体的に嵩高いA20.1バリアントは、おそらくTcdAと宿主細胞レセプターとの相互作用をよりよく遮蔽するため、TcdAの中和に優れていることを示唆している(図2F)。sd5DがTcdBのトランスロケーション・ドメインに結合するcSC5D[37]を用いて同様の実験を行ったところ、リガンド型でも非リガンド型でも、cSC5DはTcdBに対する中和能を示した(図S1DおよびS1E)。
続いて、SFcα-A20.1(sdA20.1の4コピー以上)とcSA20.1FcαA20.1(sdA20.1の5コピー以上)を比較する細胞毒性中和アッセイを行った。SFcα-A20.1は、それぞれが1つのsdA20.1に融合した4つの重鎖からなる主に二量体のFcαを含み、SFcα-A20.1がsdA20.1の4つのコピーを含み、cSA20.1FcαA20.1がsdA20.1の5つのコピーを含むことを示唆している。しかし、天然に産生されるSIgAと同様に、SFcα分子のごく一部はより大きな重合体(例えば4量体や5量体)を形成している可能性がある。これらのキメラは、1〜2コピーのsdA20.1を含む変異体と比較して、中和力の増加を示し、TcdAを標的とするキメラSIgAベースの抗体では、sdAbの数を増やすことが機能的に有利であることを示している(図2G)。これらのデータを合わせると、cSC中のsdA20.1の1コピーは、mFcα中のsdA20.1の2コピーと同様の防御を提供し、どちらもsdA20.1単独よりもかなり有利であることがわかる。4コピー以上のsdA20.1がSFcαまたはcSFcα抗体の形で含まれている場合、中和効力はさらに改善される可能性がある。
複数の抗原を標的とするcSIgAは、TcdAおよびTcdBの中和に相乗効果を示した。
cSCA20.1とmFcα-A20.1の中和力が類似していることから、我々は、1つの毒素上の複数のドメインを標的とする、あるいは2つの異なる毒素を標的とする二重特異性cSIgAを作製することにより、cSIgAの中和の幅および/または効力を増大させることができると考えた。この目的のために、我々はcSCA20.1と結合するTcdB標的Fabを探し、TcdBのグルコシルトランスフェラーゼドメイン(GTD)(毒素の主要な酵素部分)と結合することでTcdBを中和することが報告されているPA41を選択した(図3A)[38]。TcdAまたはTcdBのいずれかを含む中和アッセイにより、mIgA-PA41はTcdB(既知の抗原)と、より低い程度ではあるがTcdAの両方を中和することが明らかになった(図S2A)。TcdAの予想外のPA41依存的中和は、TcdAとTcdBのGTDドメインおよびPA41結合部位の配列類似性から生じたと考えられる(図S2B)。
図3:
図をダウンロード
新しいタブで開く
図3:
複数の抗原を標的とするcSIgAは、TcdAとTcdBの中和に相乗効果を示す。(A)TcdBのドメイン構成(上)とTcdBのGTDドメイン上のPA41-Fabの結合部位を示す(PDB: 5VQM and 6OQ5)。(B)SC、cSCA20.1、mIgA-PA41、SIgA-PA41、SIgA-STA121およびcSA20.1IgA-PA41を含む細胞毒性アッセイに使用した抗体の模式図。(C)二重特異性抗体cSA20.1IgA-PA41を含む標記分子によるTcdA(50pM)の濃度依存的中和。(D)二重特異性抗体cSA20.1IgA-PA41を含む標記分子によるTcdA(50pM)とTcdB(4pM)の濃度依存的中和。(E)二重特異性抗体cSA20.1IgA-PA41を含む標記分子によるTcdB(4pM)の濃度依存的中和。図S2および図S3も参照。
そこで、mIgA-PA41、SIgA-PA41、cSA20.1IgA-PA41およびコントロール抗体を作製し(図3B)、Vero細胞を用いた細胞毒性アッセイでTcdA(50pM)、TcdB(4pM)、またはTcdA(50pM)+TcdB(4pM)に対する中和能を測定した。PA41とA20.1を1分子中に含めることで、TcdA抗原上の2つの異なるエピトープ(GTDとCROPs)およびTcdB抗原上の1つのエピトープ(GTD)に対する二重特異性cSA20.1IgA-PA41の中和能を試験した。
注目すべきことに、TcdAのみを含むアッセイにおいて、cSA20.1IgA-PA41は、cSCA20.1またはPA41のみを組み込んだキメラ分子と比較して、TcdA(50pM)の中和効力を増強することが中和曲線から明らかになった(図3C)。続いて、TcdAとTcdBの両方を含むVero細胞細胞毒性アッセイでcSA20.1IgA-PA41の中和力を試験したところ、cSCA20.1またはPA41を単独で組み込んだタンパク質や複合体と比較して、TcdA(50pM)とTcdB(4pM)の中和力が増強されていることも明らかになった(図3D)。さらに、cSA20.1IgA-PA41とSIgA-PA41は、TcdB(4pM)に対して同程度の中和力を示したが、これはおそらくcSCA20.1がTcdBを中和せず、追加の結合部位が重要でないためであろう(図3E)。
同様の実験を、TcdA(50pM)、TcdB(4pM)、TcdA(50pM)+TcdB(4pM)に対する二重特異性抗体cS5DIgA-Pa41を用いて行った。ここで、cS5DIgA-Pa41はTcdBの2つのエピトープ(GTDとトランスロケーションドメイン)とTcdAの1つのエピトープ(GTD)に対して中和能を示した。これらの場合、cS5DIgA-PA4はすべての症例で中和の増強を示した(図S3AからS3D)。
これらの結果から、cSCをIgA Fabsとともに利用することで、2つの異なるエピトープ結合特異性を単一の二重特異性cSIgAに融合することができ、中和を増強する好ましい相乗効果をもたらすことが示された。C. difficile毒素を標的とする抗体の文脈では、これらの結果は、cSIgAにPA41-Fabを使用することで、複数の毒素に対する呼吸の増加、および1つの毒素に対する効力の増加が得られることを示唆している。
cSCはC. difficileを標的とするSIgAの可視化を容易にする。
可溶性C. difficile抗原に対するcSCとcSIgAの中和能を示した後、我々はSIgAとC. difficile表面抗原との相互作用を調べようとした。前述のように、SIgAのエフェクター機構は多様であり、多くの変数に依存しているため、SIgAが病原体のさまざまな段階にどのような影響を及ぼすかを理解することは、CDIやその他の粘膜疾患を理解し治療する上で極めて重要である。これを達成するために、我々は細菌の最外層を形成し、宿主細胞の接着に主要な役割を果たすC. difficile Surface layer protein(SLP;遺伝子SlpA)を標的とする抗体に焦点を当てた(図4)[39],[40],[41]。成熟したSLPは、高分子量(HMW)と低分子量(LMW)のSLPサブユニットが互いに非共有結合して構成され、細胞壁の上の細菌表面で準結晶配列として組み立てられている(図4)[40]。HMW-SLPは異なるC. difficile株間で高度に保存されている一方、LMW-SLPはC. difficileの血清型によって多様性を示す[42],[44]。ヒトの宿主がC. difficile SLPに対する免疫応答を起こすことを示唆する証拠がある[28],[42],[45]。例えば、SIgAは、個々の細菌を被覆したり、鎖状増殖や古典的凝集によって複数の細菌を架橋したりする可能性があり、細菌の増殖を制限したり、宿主細胞への接着を制限したり、他の下流の機能に影響を与えたりする可能性がある。このことを調べるために、SLPの低分子量部分(LMW-SLP)と結合することが報告されているsdAb、sdVHH5(別名sdCD5SLP)を選んだ[46]。我々は、sdCD5SLP を cSCCD5SLP としてリガンド型と非リガンド型に、また Fcα 融合を mFcα-CD5SLP として cSC に組み込んだ。sdCD5SLP、cSCCD5SLP(非リガンド)、cSCD5SLPFcα(リガンド)、mFcα-CD5SLPと組換えLMW-SLPの抗原結合を分析SECで確認した(図4Bと4C;図S4BからS4E)。
図4:
図をダウンロード
新しいタブで開く
図4:
SC-D2はmFPによるドメイン置換に耐性があり、抗原特異的イメージング用の高分子蛍光抗体の作製に使用できる。
(A)単分散ピークを示すcSCmCherryのSECクロマトグラム、1μM cSCmCherryの吸収スペクトル、および560nmで励起したときのcSCmCherryの蛍光スペクトル。(B)SlpAが切断され、高分子量(HMW)と低分子量(LMW)の断片を形成した後の表面層タンパク質の集合体を示す模式図。(C)mFcα-CD5SLPのLMW-SLPへの結合を示す分析的SEC。(D) 上部に示した分子とインキュベートしたSLPコーティングラテックスビーズの明視野および蛍光顕微鏡像。スケールバーは各画像の左上隅に示し、150μmである。図S4も参照。
SLPに結合するcSIgAの変異体を工学的に作製するという観点から、実験的モデル系において、コーティング、マイクロスケール架橋、古典的凝集、あるいは連鎖成長などの異なるIgAベースのエフェクター機構を区別できるような修飾を導入することも目指した。SCのD2を機能的なsdAbで置換できることを証明したことから、私たちは、他のタンパク質やドメインがD2を置換し、蛍光などの検出機能を付与できるのではないかと考えた。抗体、典型的にはIgGは、蛍光顕微鏡、蛍光アシストセルソーティング(FACS)、フローサイトメトリー、ELISAを含む蛍光ベースのアッセイを容易にするために、蛍光色素と頻繁に結合している。そこで、原理実証として、SC D2を単量体蛍光タンパク質mCherry(cSCmCherry)またはBFP(cSCBFP)で置換したcSCを作製した。
精製されたタンパク質は単分散であり、期待される吸光度および蛍光スペクトルの特性を保持していた(図4Aおよび図S4A)。これらのデータは、配位子のないcSCがD2をmFPで置き換えることに耐えることができ、広大な粘膜環境においてSIgAの検出を増強する手段を提供することを示している。
cSCmFPをリガンドcSC(cSIgA)に組み込むことができるかどうかを調べる目的で、抗原結合特異性とmCherryの異なる組み合わせを含むcSCとcSIgAのライブラリーを作製し、これらはすべて単分散複合体として精製することができた。SLP特異的蛍光であるcSmCherryFcα-CD5SLPがSLPと結合し、蛍光シグナルを与える能力を調べるため、SLPをNHS-アガロースビーズに固定化し、cSmCherryFcα-CD5SLPの存在下または非存在下でビーズを可視化した。その結果、cSmCherryFcα-CD5SLPの存在下で、SLP-アガロースビーズ上に強いmCherryシグナルが認められた(図4D)。これらの結果は、cSCmFPがcSIgA/cSFcαに効果的に組み込まれ、C. difficile SLPを含むSIgA-抗原複合体のイメージングおよび/または定量化に使用できることを示している。
SLPを標的とするcSIgAはC. difficileの架橋を促進する
我々のcSIgAの機能性を検証した後、C. difficile(ATCC-9869)の培養を、sdCD5SLP、mFcα-CD5SLP、SFcα-CD5SLP、cSmCherryFcα-CD5SLP、cSCD5SLPFcα-CD5SLP、およびcSmCherryIgA-STA121(非結合対照抗体)を含む抗体の存在下または非存在下で接種し、36時間の縦断実験を行った。光学密度(OD600nm)を用いて培養の成長をモニターし(図5A)、共焦点蛍光顕微鏡を用いて架橋を定性的にモニターし、フローサイトメトリーを用いて架橋を定量化した。
図5:
図をダウンロード
新しいタブで開く
図5:
高分子抗SLP抗体はC. difficileの架橋を引き起こす。
(A) キーに示したサンプルのOD600nm対時間。太い縦格子線は、フローサイトメトリーと画像化のためにサンプルを採取した時点を示す。三角形は、凝集イベントの数がピークに達した時点を示す。(B) 未処理のC. difficileと指定した抗体またはコントロールで処理したC. difficileについて、合計100,000イベントのうち記録されたクランピングイベントの平均数を示す。(C)8時間後の時点における標記抗体またはコントロールで処理したC. difficileのフローサイトメトリー解析と、それぞれの共焦点顕微鏡像。スケールバーは50μm。図S5および図S6も参照。
未処理のC. difficile、cSmCherryIgA-STA121(コントロール)、およびcSCCD5SLPで処理したC. difficileは最初から急速に増殖し、約1時間後に対数相に入り、約6時間後にOD600のピークに達し、その後約6時間から36時間までOD600は着実に減少した(図5A)。未処理、cSmCherryIgA-STA121処理、またはcSCCD5SLP処理培養とは対照的に、sdCD5SLP、mFcα-CD5SLP、SFcα-CD5SLP、cSmCherryFcα-CD5SLP、およびcSCD5SLPFcα-CD5SLP処理培養は、対数期および定常期(約8~14時間)に続いてOD600の急速な減少を示し、その後OD600は一定であった(図5A)。このことは、cSCCD5SLPが標的抗原にin-vitroで結合しても(図5A)、増殖しているC. difficileに顕著な影響を与えないことを示していると思われる。
同時に、2時間、4時間、6時間(対数期)、8時間、10時間、15時間(定常期および/または死滅期)に採取したサンプルをフローサイトメトリーと共焦点顕微鏡を用いて分析した。フローサイトメトリーデータは、0時間の未処理のC. difficileを基準に最適化され、ゲートが設定された。ゲート内のイベントは、C. difficileの短鎖からなる正常な細胞集団とみなされた。このゲートの外側のイベントは、より高いFSC-AとSSC-Aに対応し、サイズと複雑さの増加を示し、凝集細胞と分類された。100,000イベント中のクランピングイベント数を記録し、経時的な値の変化を解析した(図5B)。
塊状化は、mFcα-CD5SLP、SFcα-CD5SLP、cSmCherryFcα-CD5SLP、cSCD5SLPFcα-CD5SLPなど、CD5SLPを少なくとも2コピー含むキメラ抗体で処理したC. difficileサンプルで検出された。未処理のサンプルと、CD5SLP、cSmCherryIgA-STA121、cSCCD5SLPまたはsdCD5SLPを0から1個含むサンプルは、凝集イベント数の緩やかな増加を示し、約3500イベントで停滞した(図5B)。このことから、増殖中のC. difficileでは、細胞分裂、混雑、非特異的相互作用の結果として、約3500回の凝集イベントが予想され、CD5SLPの単一コピーを持つ抗体の存在によって凝集に顕著な影響はないことが示された。対照的に、mFcα-CD5SLP、SFcα-CD5SLP、cSmCherryFcα-CD5SLP、およびcSCD5SLPFcα-CD5SLPは、ほとんどのサンプルで最大の凝集が観察された8時間の時点で、各サンプルについてそれぞれ9217、17764、17312、14656の凝集イベントを示した(図5B)。SFcα-CD5SLP、cSmCherryFcα-CD5SLP、cSCD5SLPFcα-CD5SLPが4時間後に3500イベントを超えるのとは対照的に、mFcα-CD5SLPは6時間後に3500イベントを超える。
フローサイトメトリーに使用したサンプルの共焦点イメージングでは、少なくとも2つのCD5SLPを持つ抗体で処理した培養での凝集が定性的に示され、フローサイトメトリーの結果を検証した(図5C; 図S5A)。これらのデータを総合すると、CD5SLPの少なくとも2つのコピーが凝集を誘導するのに必要であるが、1つの抗体にCD5SLPが4つ以上含まれると凝集が促進されることが示された。これらの結果は、蛍光顕微鏡画像を用いて成長を分析し、架橋を定量化した再現実験と一致していた(図S6AからS6C)。さらに、共焦点顕微鏡と比較して、フローサイトメトリーを用いた架橋定量化では、CD5SLPを2コピー含む抗体と4または5コピー含む抗体との間の凝集の違いが検出されたが、これは代表的な共焦点画像で細胞をカウントしたときには観察されなかった(図S6B)。これは、フローサイトメトリーによる培養全体の定量とは対照的に、培養サンプルから採取したアリコートを用いたことに一因があるかもしれない。さらに、フローサイトメトリーで検出される塊の大きさは、最初の共焦点顕微鏡実験で「塊状」と判定されたものよりも大きい可能性が高い。注目すべきことに、cSmCherryFcα-CD5SLPは、mFcα-CD5SLP、SFcα-CD5SLP、cSCD5SLPFcα-CD5SLPのような他のキメラと区別できない塊状化を示した。
異なる時点のcSmCherryFcα-CD5SLP処理培養に対応する共焦点顕微鏡画像を観察すると、C. difficile細胞は早い時点(2時間)から抗体でコートされていた。2-6時間(対数期)の間、画像はcSmCherryFcα-CD5SLPで均一にコートされたように見える細胞の塊を示した。しかし、8時間前後(定常期)には、培養中のC. difficile細胞の一部で不均一な抗体コーティングが観察され、これは典型的な棒状構造ではなく「破裂」したような外観を特徴とする形態学的変化と相関していた(図6)。また、sdCD5SLP、mFcα-CD5SLP、SFcα-CD5SLP、およびcSCD5SLPFcα-CD5SLPで8~36時間処理した培養物では、程度の差こそあれ破裂した形態現象が観察されたが、cSCCD5SLPでは36時間後にのみ最小限の破裂が観察された(図S7)。未処理およびcSmCherryIgA-STA121処理培養では、破裂形態は観察されなかった。このことは、増殖しているC. difficile SLPを標的とする抗体は微生物に形態変化を誘導することができ、我々の実験ではエフェクター細胞がないにもかかわらず、病原体に長期的な破壊的影響を与えることができることを示している。
図6:
図をダウンロードする
新しいタブで開く
図6:
cSmCherryFcα-CD5SLPはC. difficileをコートし、経時的な形態変化を引き起こす。
示したサンプルのフローサイトメトリーデータと関連する共焦点蛍光顕微鏡画像から、時間経過に伴う凝集イベントの数と赤色蛍光シグナルが明らかになった。すべての画像のスケールバーは25μmである。図S7も参照。
二重特異性cSIgAはC. difficileの凝集を促進し、細胞毒性を低下させる
SLPを標的とするSIgAがC. difficileの凝集を促進することを決定し、cSIgAが毒素を中和することを決定したので、mFcα-CD5SLPとcSC5D(TcdBを標的とする)を含むcS5DFcα-CD5SLPと呼ばれる二重特異的cSIgAを作製した。事前の実験と一致して、cS5DFcα-CD5SLPを含む培養は、凝集と最終的な細胞破裂を示した(図7A-C)。続いて、cSC5Dを欠き野生型SCをコードするcS5DFcα-CD5SLPまたはSFcα-CD5SLPで処理した培養物の上清の細胞毒性をVero細胞細胞毒性アッセイで試験した。未処理のコントロールと比較して、cS5DFcα-CD5SLPまたはSFcα-CD5SLPで処理した培養の上清は細胞毒性の減少を示し、cS5DFcα-CD5SLPで処理した培養は細胞毒性の最大の減少を示した(図7B)。これらのデータは、CD5SLPとcSC5DをcSIgAに含めることで、培養微小環境におけるTcdBの毒性を軽減できることを示唆している。cSC5Dの中和能を試験した実験で観察されたように、cSC5Dが直接細胞毒性を低下させると予想されるが、CD5SLPが細胞毒性を低下させる根本的なメカニズムはあまり明確ではない。SLP標的抗体の存在下でC. difficileの増殖が全体的に減少するためかもしれないし、毒素産生を低下させる遺伝子発現の変化のためかもしれないし、その両方の組み合わせかもしれない。どのようなメカニズムにせよ、SLPを標的とするSFcα-CD5SLPは、毒素産生など他の病原性因子に間接的に影響するようであり、天然に存在するSLPに対する抗体は、病原性と上皮細胞障害を複数の方法で軽減するのに役立つ可能性が示唆された。この結果は、SLP標的抗体や二重特異性抗体が、病原体の病原性の複数の側面に対抗し、細胞毒性作用を最小化する治療の可能性をも示している。
図7:
図をダウンロード
新しいタブで開く
図7:
C. difficile SLPと毒素を標的とする二重特異性抗体は、細胞毒性を低下させると同時に凝集を引き起こす。
(A)抗SLP抗体SFcα-CD5SLP、およびSLPとTcdBを標的とする二重特異性抗体cS5DFcα-CD5SLPの存在下で増殖したC. difficileの増殖曲線。(B) 示した抗体で処理した培養から得られた上清の細胞毒性。(C) 示した時点でcS5DFcα-CD5SLPで処理したC. difficile培養のフローサイトメトリーおよび蛍光顕微鏡画像。すべての画像のスケールバーは25μmである。
考察
ヒトはSIgAの産生に多大なエネルギーを費やしているが、SIgAのユニークなエフェクター機構と多様な粘膜抗原への影響を支配する分子機構はまだ十分に解明されていない。我々の研究から、SIgAは大規模な構造改変に対して驚くほど寛容であることが明らかになり、その結果、そのエフェクター機構と治療薬としての開発可能性を探ることができた。我々は、SIgAに依存する抗原中和と免疫排除のエフェクター機構に着目した。この機構は、免疫エフェクター細胞や炎症が制限されている粘膜分泌液において支配的であると考えられている[11], [47]。これらのメカニズムは、SIgAのユニークな分子構造によって支えられていると推定されている。SIgAの低温電子顕微鏡構造とFabsのモデリングによって明らかにされたこの構造は、コンフォメーション的に非対称であり、SCと2つのFcR結合部位が立体的にアクセス可能なままである一方で、Fabsが占めることのできる潜在的な位置を制約している可能性が高い[17],[30]。このような幾何学的制約は、SIgAの抗原や他の因子への結合に独特な影響を与え、おそらく高貪食性相互作用を促進することで、ある因子との相互作用を有利にし、かつ/または他の因子と結合するために最適な間隔をとるという仮説が立てられている[17]。SIgAの典型的な高分子配列は、単量体抗体と比較して架橋の利点を提供する、あるいはSIgAが与えられた抗原に結合すると(コーティングとは対照的に)架橋を誘導するとしばしば推定される。しかし実際には、このことが実験的に実証されたり定量化されたりしたことはほとんどなく、宿主や微生物への影響も解明されていない。例えば、ヒトはC. difficile SLPに対するSIgAを開発するが、抗原との結合が、コーティング、ナノスケールの架橋(1つの微生物上のSLP間)、微生物スケールの架橋、あるいはその他の効果をもたらすのかどうか、また、そのSIgAが単量体抗体と比較して優位性をもたらすのかどうかは、未知のままである。ここで我々は、これらおよび関連する疑問に取り組み、(1) IgA上の同一のエピトープ結合部位の数を増やすと、可溶性抗原の中和が促進されること(例:TcdA)(2) 単一のIgA上に異なるエピトープ結合部位を組み合わせると、複数の可溶性抗原の中和が促進されること(例:cSA20によるTcdAとTcdB。 1IgA-PA41)(3)単一のIgA上の同一のエピトープ結合部位の数を増やすことで、微生物の架橋や標的細胞の破裂などの免疫排除機構を強化することができる(4)単一のcSIgA上の異なるエピトープ結合部位を組み合わせることで、複数の病原性因子エピトープを標的とする(例. 5)単一のcSIgAにエピトープ結合部位とmFPを組み合わせることで、cSIgA-抗原相互作用の可視化と定量化、および架橋などの関連する結果を容易にすることができる。これらの結果は、単量体抗体に対するSIgAの優位性を示し、天然のSIgAエフェクター機構、特にCDI対策に関連する機構を理解するのに適用でき、SIgAの有益な特性を強化できることを示している。
我々がC. difficile抗原とSIgAとの相互作用に焦点を当てているのは、SIgAがCDIに与える影響についての理解が限られていること、また、不完全な病原体除去、疾患の再発、抗生物質耐性を持つヒト病原体の増加など、抗生物質による治療には多くの課題があることに起因している。しかし、ベズロトクスマブは静脈内投与されるため[26]、上皮バリアが破壊された後にTcdBを標的としているのに対し、天然または人工のSIgAは粘膜内のC. difficile抗原を標的とし、損傷が起こる前に感染を抑える可能性がある。実際、われわれのデータは、毒素中和の場合、結合部位の数を増やすことが有利であり、sdA20.1のコピーを4〜5個含むSIgAおよびcSIgAは、sdA20.1のコピーを1〜2個含む変異体と比較して、中和力が増大することを示している。予想外なことに、cSCA20.1はTcdAを中和できたが、sdA20.1単独では中和できなかった。sdA20.1はTcdAのCROPドメインを標的としているが、CROPは唯一の受容体結合領域ではなく、CROPを欠く毒素もある程度の細胞毒性を維持している[48],[49]。したがって、cSCA20.1はCROPに結合すると同時に、隣接する受容体結合部位を立体的に閉塞している可能性がある。この発見は注目すべきものである。というのも、1つの宿主レセプター結合部位(例えばsdA20.1結合部位)をブロックすることが中和メカニズムの重要な一部であるように見える一方で、感染という状況において中和を達成するためには他の因子が必要であることも示唆しているからである。注目すべきことに、初乳SCはC. difficileの毒素の影響を制限することに関与している[18]。この作用の根底にある生得的な様態のメカニズムは、糖鎖を介するものであることが決定されている[18]。我々は、sdA20.1(または他の毒素結合可変ドメイン)を複数コピー含むSIgAまたはcSIgAの場合、SIgAのサイズがレセプター結合を阻害するのに役立つだけでなく、SIgA依存性の毒素架橋と多毒素可溶性凝集体の形成により、抗体が直接標的としない部位を効果的にブロックすることで、宿主細胞レセプターへの毒素のアクセスがさらに制限されるのではないかと推測している。さらに、我々の実験では、1つのcSIgAが複数の毒素を標的とする場合、相乗効果も観察された。多毒素凝集体の形成、および/または1つの毒素エピトープに結合したcSIgAが他の毒素のレセプターとの結合をブロックする能力は、我々が観察した相乗効果を説明するかもしれない。複数の毒素を発現するC. difficile株(超蔓延性C. difficileリボタイプBI/NAP1/027など[50])の場合、cSIgAが治療の利点をもたらす可能性がある。
C. difficile毒素はCDIに関連する上皮細胞損傷の大部分に関与している;しかしながら、微生物の持続的増殖は再発の主な要因である。ヒトはC.difficile表面抗原に対する抗体を産生するが、これらの抗体がC.difficileを上皮バリアから効果的に排除し、増殖を制限し、および/または排出を促進するかどうかについては研究されていない[28]。先行研究では、サルモネラ・チフスムリウム(STm)表面抗原を標的とするSIgAが、連鎖増殖および古典的凝集機構を介して免疫排除を促進し、また水平遺伝子移入を制限し、および/または微生物の表面を変化させることができることが示された[16],[17],[51],[52]。我々の実験から、SLPを標的とする抗体は細胞の生存率に影響を与えることができ、mFcα-CD5SLP、SFcα-CD5SLP、cSmCherryFcα-CD5SLP、cSCD5SLPFcα-CD5SLPのような2つ以上の抗原結合部位を持つ抗体は、未処理の対照、陰性対照抗体、sdCD5SLPやcSCCD5SLPのような単一のSLP結合部位を持つ抗体と比較して、C. difficile細胞の架橋を効果的に誘導できることが明らかになった。注目すべきは、4つ以上の抗原結合部位を持つものは、さらに大きな架橋能を示したことである。このことは、自然発生的な腸内感染において、SLPを標的とするSIgAは、免疫排除メカニズムを介してC. difficileを効果的に制限する可能性が高いことを示している。我々のデータは、架橋の前にSIgAが細菌細胞をコーティングし、その後の架橋が高い細胞密度と相関することを明らかにした。このことは、SIgAがある微生物表面のSLPに結合しても、別の微生物表面への結合は妨げられないことを示唆しており、古典的凝集が支配的なメカニズムであることを示唆している。古典的凝集は、密度が高くなるにつれて増加する細胞間衝突の確率によって駆動されるが、これとは対照的に、連鎖成長は細胞密度が低くても起こりうる。私たちが観察した架橋の何割かは連鎖成長が寄与している可能性を否定することはできないが、細胞密度が高い場合に起こる架橋に加えて、未処理のC. difficile細胞は棒状で、しばしばペアや短い連鎖で現れることが知られているのに対して、私たちの画像では、端から端までつながった長い連鎖細胞ではなく、様々な向きの塊状の細胞が観察されたことに注目したい[53]。自然発生する感染とそれに伴う宿主の抗体反応との関連において、SIgA-SLP依存的な凝集は、おそらく上皮細胞からの細菌の排除を促進し、蠕動運動による除去を促進するであろう。興味深いことに、我々のデータは、抗SLP抗体への長期暴露がC. difficile細胞の破裂と死を促進することも示している。このC. difficileの破裂は、sdCD5SLPや、より少ない程度ではあるがcSCCD5SLPも細胞の破裂を引き起こしたことから、架橋とは無関係のようである。
我々の研究はSIgAのエフェクター機構に関する新しい洞察を提供したが、同時にSIgAはその有益な特性を増強するために改変することができるという証拠も提供した。2つの可溶性C. difficile毒素、TcdAとTcdBを標的とするcSIgAの場合、それぞれを標的とする場合と比較して、相乗効果が観察された。具体的には、cSA20.1IgA-PA41はTcdAの中和を増強し、TcdAとTcdBの両方の毒素を中和した。また、TcdAとSLP結合能を一つのSIgAに結合させることで、C. difficile細胞の架橋を促進し、細胞毒性を軽減できることもわかった。驚くべきことに、TcdAを中和するcSCA20.1の能力ではなく、sdA20.1の能力によって証明されるように、sdAbsよりもcSCAの方が優れていることも観察された。哺乳類は大量の遊離SCを粘膜に分泌しており、その理由はまだ不明であるが、遊離SCはC. difficileを含むいくつかの微生物に対して生得的な防御を提供している可能性がある[18],[12]。SCは粘膜分泌液中で安定であると考えられている。SCは複数のN-結合型糖鎖を含み、コンパクトな閉鎖構造をとっており[29]、組み込まれたsdAbの活性、安定性、あるいは局在性(非特異的結合など)を高める可能性があると推測される。しかしながら、cSCCD5SLP は sdCD5SLP と比較して、死相を誘導することも、細胞の破裂を促進することも できなかったので、SC が常に有利に働くとは限らないことに注意したい。いずれにせよ、我々のデータは、操作されたSIgAがCDIのような感染症を治療する可能性があるというエキサイティングな可能性を強調している。我々は、cSIgA生物製剤を感染部位(例えば、大腸や結腸)に投与することで、高リスク者の疾病を治療したり、疾病を予防したりすることを想定している。
ここでは、SIgAのエフェクター機構と標的C. difficile抗原のサブセットに焦点を当てたが、より広範には、複数のsdAbまたはmFPがSC-D2に取って代わることができることを示した我々の結果は、事実上あらゆるsdAbまたはmFPがSC-D2に取って代わることができることを示しており、SCまたはSIgAを追跡し、および/またはそのエフェクター機構を強化する様式を提供する。このことは、cSCを異なるdIgAと安定的に共発現させる我々の能力と相まって、複数の抗原結合部位および/または異なる抗原結合部位および/または検出方法を組み合わせたcSIgAの多様なライブラリーを作製する可能性を示している。例えば、我々はcSCmCherryとcSCmBFP dIgAを共発現させ、cSmFPIgAを形成した。このcSmFPIgAは、Fabsの機能を損なうことなく、またSIgAの全体的な構造やコンフォメーションの利点を変えることなく、フローサイトメトリーによる抗原特異的イメージングと定量化に利用することに成功した。また、cSIgAはSIgAのFcR結合部位を変化させることはないと予想され、原理的にはFcR依存的なメカニズムの研究に用いることができる。我々は、cSmFPIgAがSIgA-蛍光色素結合体よりも有利であり、ミリグラムの量が必要とされるであろう抗体の粘膜投与(例えば、マウスの腸への投与)を含む実験において、ネイティブなSIgAと導入されたcSIgAを容易に区別する能力を提供することを想定している。また、cSmFPIgAを用いることで、微生物へのSIgAのコーティングや架橋の定量化(例えば、フローサイトメトリーや1分子蛍光を用いる)が簡便になり、SIgAのエフェクター機能や治療の可能性についての理解を深めることができるようになると考えている。
材料と方法
構築物の設計とクローニング
ヒトJCのコード領域(UniProt-P01591、ネイティブシグナルペプチド配列)をコドン最適化、合成し、マウスJC構築物と同様にpD2610v1(ATUM)にクローニングした[30]。ヒトIgA2m1発現コンストラクトは、TPAシグナルペプチド(残基MDAMKRGLCCVLLCGAVFVSPAGA)とともにそれぞれの可変ドメイン配列を定常ヒトIgA2m1 HC(AAT74071.1)[55]に融合し、pD2610v13(ATUM)に挿入することにより作成した。簡単に言うと、異なる抗体のVH領域遺伝子セグメントを選択し、コドンを最適化し、合成し(IDT)、Overlap-extension PCR[56]を用いてIgA2m1定常重鎖ドメインと融合させ、得られた融合体をpD2610v13(ATUM)にElectraクローニングした。Fc融合sdAbバリアントは、sdAbをコードするセグメントをIgA2m1のCH2-CH3ドメインに融合させた類似の方法で作製した。Hisタグ付きFcαは、PCRを用いてTPAシグナルペプチドとHHHHHGS配列をIgA2m1 CH2-CH3配列に融合することにより作製した。得られた遺伝子セグメントは、Electra-cloning(Atum)を用いてpD2610v13ベクターにクローニングした。VLドメインをヒトIgLambda-1(UniProt-P0DOX8)のCLドメインと融合させ、シグナルペプチド配列(残基METDTLLLWVLLWVPGSTG)を付加し、ElectraクローニングによりpD2610v13ベクターに挿入した。Secretory Component (SC)(UniProt-P01833)と呼ばれるヒト高分子免疫グロビン受容体(pIgR)のエクトドメイン領域も同様に合成し、コドンを最適化し、Electraクローニングを用いてpD2610v1とpD2610v13の両方にクローニングした。D2を置換するキメラSC(cSC)は、キメラ部分をコードし、D1およびD3と重複する配列を持つコドン最適化合成DNA(IDT)を用いて、オーバーラップ-エクステンションPCRにより作製した。得られた遺伝子セグメントは、Electra cloning (Atum)を用いてpD2610v1またはpD2610v13ベクターにクローニングした。sdAbsをコードするコンストラクトは、N末端シグナルペプチドとC末端ヘキサヒスチジンタグを公表されているVHH配列に融合させることにより作成した。この配列はコドンを最適化して合成し、pD2610v13ベクターにElectraクローニングした。異なるVH、VL、VHHを以下の表1.1に示す。
インラインで見る
表1.1
抗体情報
トランスフェクションおよび採取
ExpiFectamine 293 transfection kit (ThermoFisher: A14525) を用いて、全てのコンストラクトをExpi293F (Gibco: A14527) 細胞に一過性にトランスフェクション (SC, cSC, sdAb, mFcα-fused-sdAb) または共トランスフェクション (SFcα, SIgA, cSFcα, cSIgA) した。トランスフェクションの4~5日後に上清を回収し、0.22μm PESボトルトップフィルター(Millipore Sigma)でろ過した。
細菌タンパク質の発現
TXA1(pET28aベクター)およびSLP(2BCTベクター)の発現用プラスミドは、Shannon Sirk博士の研究室(UIUC)から提供された。プラスミドをBL21大腸菌に形質転換し、適切な抗生物質耐性でスクリーニングした。それぞれのコロニーを3mlのスターター培養液に接種し、BL21 (C2530)のNew England Biolabs (NEB)のプロトコールに従ってIPTG誘導を行い、タンパク質を生産するために100-250mlの大培養液に使用した。37℃で4時間のタンパク質発現後、細胞を4000xgでペレット化した。このペレットを、リゾチーム、DNAse、プロテアーゼインヒビターカクテル(Pierce)を含む溶解バッファー(50mM Tris 7.5, 500mM NaCl, 5-10% glycerol, 10mM Imidazole)に懸濁した。再懸濁したペレットを、混合物が水のような粘性になるまで超音波処理した。この溶解液を10000xgで45分間遠心分離し、細胞残屑を除去した後、上清をNi-NTAアフィニティー樹脂(Qiagen: 30210)を用いて細菌タンパク質を精製した。
タンパク質の精製
トランスフェクション後に回収した上清または細菌細胞溶解液を、適切なアフィニティー精製を用いて抗体/タンパク質の精製に使用した。SC、cSC、sdAbおよび細菌細胞溶解液からのタンパク質(SLPおよびTXA1)は、トリス緩衝食塩水(TBS:20mM Tris+150mM NaCl、pH=7.4)+250mMまたは500mlイミダゾールを溶出に用いて、Ni-NTA樹脂(Qiagen:30210)で精製した。同様に、His-Fcαコンストラクトと複合体は、Ni-NTA精製法とTBS+250mM、TBS+500mMイミダゾール、TBS+750mMイミダゾール、TBS+1Mイミダゾールを用いた段階溶出勾配により精製した。Fc融合抗体、全長抗体、およびそれぞれのcSCとの複合体は、Capture Select IgAアフィニティー樹脂(Thermo Scientific: 194288010)を用いて精製し、製造元のプロトコールに従って、pH=3で0.1Mグリシンを用いて溶出した。精製後、全てのタンパク質を1Xリン酸緩衝生理食塩水(PBS) pH=7.4 (Cytiva)中で適切なMWカットオフのAmicon遠心濃縮機(Millipore Sigma)を用いて緩衝液交換し、最終容量0.5-1mlに濃縮した。PBS中のタンパク質は、サイズ排除クロマトグラフィー(SEC)の前に、スピンフィルター(Millipore Sigma)を用いて0.22umでろ過した。タンパク質サンプルのSECは、Superose 6カラム(Cytiva社製)またはSD200 10 300(Cytiva社製)カラムを使用し、AKTA pure(Cytiva社製)を用いて自動平衡化、サンプルロード、500ul溶出液へのサンプル溶出、およびそれぞれの吸光度データ収集を行った。SEC後の溶離液は、使用するまでPBSに保存した。
分析SEC
タンパク質-タンパク質ペアの等モル比を計算し、推定分子量に基づいて、ProtParamウェブサイト(https://web.expasy.org/protparam)から決定した。実験には最低50ugの各タンパク質を使用した。計算に基づいて、等モル量のタンパク質-タンパク質混合物を、最終容量1mlの1X PBSに加え、室温で1時間インキュベートした後、SECの前に4℃でインキュベートした。コントロールは、混合物と同じモル量の個々のタンパク質を1mlのPBSに入れたものである。タンパク質-タンパク質混合物とコントロールは、AKTA pure(Cytiva)にSD200 10 300カラム(Cytiva)を取り付けてSECにかけた。それぞれのクロマトグラムは、Unicorn 7.5評価ソフトウェア(Cytiva)からエクスポートし、OriginPro 2020グラフ作成・分析ソフトウェア(OriginLab: https://www.originlab.com/)を使ってプロットした。
蛍光および吸光度測定
cSCmCherryまたはcSCBFPを含むキメラ蛍光タンパク質について、蛍光および吸光度スペクトルの測定は、平坦な透明底96ウェルプレート(Thermo Scientific: 165305)に100ul容量のPBS中1uM cSCを用いて行い、BioTekのCytation 5プレートリーダーを用いて吸光度および蛍光スペクトルを測定した。吸光度は適切な範囲で測定し、蛍光は cSCmCherry は 586nm 励起、cSCBFP は 400nm 励起、適切な発光範囲で測定した。バックグラウンドサブトラクションにはPBSの吸光度と蛍光スペクトルを用いた。
NHS-SLPビーズの形成
乾燥Pierce™ NHS活性化アガロースとPBS中の精製表面層タンパク質(SLP)をSLPコーティングアガロースビーズの作製に用いた(Pierce NHS活性化アガロースのプロトコールに従う)。1.5mlのエッペンドルフチューブにNHS活性化アガロースを10-50ug(ウェット量に換算すると75-375ug)入れた。これにPBS中の1uM表面層タンパク質(SLP)1mlを加え、結合プロセスを開始した。結合は、4℃、100rpmで穏やかに振盪することにより行った。NHS-アガロース-SLP混合物を1000xgで遠心し、上清を除去した後、同様に1000xgで遠心して1mlのPBSで2回洗浄した。最終的なウェット量は約250ugであった。洗浄したアガローススラリーに1Mのエタノールアミン(Sigma Aldrich: E9508)をウェットボリュームの2倍量加え、100rpmで振とうしながら室温で20分間インキュベートした。この後、TBSで3回洗浄し、最終的なSLP-アガロースは実験用にTBSで最終容量1mlに保存した(アガロース濃度約250ug/ml)。
ビーズ蛍光研究
ビーズ結合およびイメージングアッセイのために、SLP結合ビーズスラリーを撹拌し、100ulのスラリーを1.5mlのラベル付きエッペンドルフチューブに分注した。スラリーを1000xgで遠心し、上清を捨て、TBS中の5uM分析対象タンパク質100ulと交換した。分析物とスラリーの混合物を室温で1時間インキュベートした後、TBSで3回洗浄し、最後に100ulのTBSに再懸濁した。この最終スラリー10-20ulを、カバースリップ(フィッシャーサイエンティフィック社製)を端に透明マニキュアでシールしたスライドガラス(フィッシャーサイエンティフィック社製)上に置いた。アガロースビーズをLeica倒立蛍光顕微鏡で明視野および蛍光イメージングした(Kai Zhang研究室)。収集した画像は、Imaris Viewerソフトウェアで可視化した。
Vero細胞細胞毒性中和アッセイ
Vero細胞(ATCC-CCL-81)を、ダルベッコ変法イーグル培地(DMEM)(ギブコ社製)+10%ウシ胎児血清(FBS)(ギブコ社製)の完全培地中で、組織培養処理した96ウェルプレート(サーモサイエンティフィック社製)に1ウェルあたり10000細胞ずつ播種し、8%CO2中、37℃で培養した(0日目)。20-22時間後(day1)、各ウェルの完全培地を除去し、完全培地中の各濃度の毒素と抗体のプレインキュベート混合物を各96ウェルに添加した。その後、プレートを65時間インキュベートした。細胞生存率を測定する前に、細胞をMEM(ギブコのアールズMEM)で少なくとも2回洗浄し、アラマーブルー細胞生存率試薬(Invitrogen: DAL1100)の1:10混合液をMEMで調製し、この混合液100ulをすべてのウェルに加えた[36]。その後、プレートを37℃で3時間インキュベートし、Biotek社のCytation 5 Plate readerを用いて、各ウェルの蛍光強度を吸光度560nm、発光590nmで測定した。TcdAアッセイには50pM濃度のTcdAを、TcdBアッセイには4pMのTcdBを、二重毒素中和アッセイには50pMのTcdAと4pMのTcdBを用いた。抗体濃度とその希釈系列は、各実験で比較するための適切な範囲のVero細胞生存率を得るために、実験要件に応じて変化させた。
C.ディフィシル中和アッセイ
Clostridiodes difficile ATCC-9689-fz(株番号-90556-M6S、トキシノタイプ0、リボタイプ001)をすべての実験に使用した。プロトコール[59]、[60]に従って、C. difficileは37℃の嫌気チャンバー(Coy Laboratories)で増殖するように最適化した。0.1%L-システイン(Sigma Aldrich: 168149)を添加した酵母エキス(5g/L)(BHIS)入り脳心筋梗塞培地(37g/L)を調製し、滅菌培養チューブに分注し、C. difficile接種前に嫌気性チャンバーで1日間予備減菌した。さらに、平底および透明底の96ウェルプレート(Fisher Scientific社製)、チップ、その他の消耗品などのプラスチック製品は、嫌気室で少なくとも1日間予備還元した。C.difficileは、公表されているプロトコール[59]、[60]に従って凍結グリセロールストックから接種した。この12~18時間の一晩培養をC. difficile中和アッセイの設定に用いた。一晩のOD600nmを測定し、それに応じてサンプルをBHIS+0.1% L-システインで推定OD0.05に希釈した。96ウェルプレート中で、希釈したC. difficileを、BHISで希釈した等量の8uM濃度の抗体と1:1の比率で混合し、アッセイにおける抗体の最終濃度を4uMとした。プレートは通気性の膜(Millipore Sigma)で密閉し、結露を防ぎ、蒸発を最小限に抑えた。フローサイトメトリーおよび共焦点顕微鏡のスライド作製のためのサンプル抽出には、プレートを2枚セットした。
C. difficile増殖上清の細胞毒性
C. difficile 上清の細胞毒性は、Cerillo Stratus プレートリーダー専用の 96 ウェルプレートで、指示された OD600nm 測定で上清サンプルを採取することにより測定した。簡単には、細胞をスピンダウンし、0.22μmのフィルターで上清を濾過してプレートに回収した。処理サンプルと未処理サンプルの上清の細胞毒性は、前述のように培養したVero細胞(ATCC-CCL-81)を用いて分析した。細胞毒性評価は、DMEM+10%FBSで2-5%上清開始濃度および各5倍希釈系列を、予め播種した10000cells/wellのVero細胞に投与して行った。細胞生存率は、前述のようにアラマーブルーアッセイを用いて48時間後に分析した。適切な陽性対照(完全培地+100pMTcdA+10pM TcdB)と陰性対照(完全培地)を用いて生存率を推定した。
C.difficileサンプルの共焦点顕微鏡観察
抗体の存在下および非存在下でのC. difficileの増殖中の異なる時点で、C. difficile培養のそれぞれの96ウェルプレートから5ulのサンプルを採取した。5ulのサンプルをPBS中の5ulの8%パラホルムアルデヒド(PFA)(Fisher Scientific)と混合し、スライドグラス(Fisher Scientific)上に置き、カバースリップ(Fisher Scientific)を鉗子を用いてゆっくりと置き、マニキュアを用いてエッジをシールした。スライドの前処理はすべて嫌気槽内で行った。ウェットマウント顕微鏡スライドは、Zeiss LSM 900を用い、63X/oil、1.4NA、1倍ズームで撮影した。C.difficileの自家蛍光は緑色チャンネルで最も高いため、488nmの励起光と0.5%のレーザー強度を用いて緑色の自家蛍光を画像化した。位相差データ収集のため、透過型光電子増倍管(T-PMT)をmCherryと同じチャンネルで561nmに設定した。緑チャンネルと赤チャンネルには適切なゲインが用いられ、蛍光が最も弱いサンプルと蛍光が最も明るいサンプルに基づいて最適化された。画像は、手作業でカウントするためのタイルとして、および/または必要に応じてZ-stackで収集した。
画像の解析
細菌の塊は、Image-J cell counter plugin(https://imagej.nih.gov/ij/)を用いて、位相差画像上の細菌を手動でカウントすることにより定量化した。計数は決められたルールに基づいて行われた: 塊状細胞は、3個以上の細胞が端から端まで(非直線状) に接触しているものと定義し、塊状でない細胞は、単細胞で分散している、 あるいは端から端まで(直線状)に成長し連結しているものと定義した。図の画像はImaris viewer(https://imaris.oxinst.com/imaris-viewer)を用いて作成した。
フローサイトメトリー
未処理または抗体処理したC. difficile培養物100ulを96ウェル培養プレートから抽出し、最終濃度2%PFAになるようにPFAと混合し、少なくとも30分間RTでインキュベートした。固定したC. difficile培養液をPBSで希釈して最小容量400ulとし、LSR Fortessaフローサイトメーターにロードした。PMTの電圧と閾値は未処理のC. difficileに基づいて調整し、FITCとTexas redの蛍光色素を設定して緑と赤の蛍光を記録した。未処理のC. difficileについて、0時間の時点で300,000イベントが記録された。未処理のC. difficileでは、0時間で300,000イベントが記録された。他のすべての時点では、処理サンプルと未処理サンプル、およびそれらの複製について、100,000イベントが記録された。20時間と36時間に採取したサンプルは、細胞破裂が優勢であったため、フローサイトメトリーでは解析しなかった。
フローサイトメトリーデータ解析
フローサイトメトリーデータ解析には、FCS Express 6および7を使用した。すべてのサンプルは、実験のT=0時間における未処理のC. difficileサンプルに従って、SSC-A対FSC-Aのドットプロットでゲートされた。T=0時間における99%以上のC. difficile細胞を囲むゲートを「正常集団」とし、SSC-A対FSC-Aの高値を考慮してこのゲートの外側に描かれた別の多角形のゲートを「塊状集団」と分類した。FCS Expressから塊状集団の総数の統計データを抽出し、OriginPro 2020を使って各サンプルについて時間に対してプロットした。SSC-A vs FSC-Aのドットプロットは、FCS Expressから画像としてエクスポートした。
著者貢献
本研究は、B.M.S.およびS.K.B.が発案し、実験はS.K.B.が実施した。すべての著者がデータ解析と原稿執筆に貢献した。
利害関係
BMSとSKBは、SIgA工学に関する特許(WO2023044419)の発明者として記載されている[54]。(https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023044419&_cid=P12-LHI6X3-04314-1)
補足図
図S1
図のダウンロード
新しいタブで開く
図S1
図2の関連図:機能的cSCは、リガンド型でも非リガンド型でも生成・利用できる。
(A)cSCA20.1、cSCA5.1、cSCB39、cSC5D、cSCCDB1LCの正規化単分散SECピーク。(B)sdA20.1、TcdAフラグメント、TXA1、およびsdA20.1-TXA1複合体のSEC溶出プロフィールの模式図と分析。分析的SECは、等モル量のTXA1とsdA20.1を室温で1時間インキュベートした後、混合物を4℃に冷やしてSECを行った。TXA1とsdA20.1をそれぞれ同様にインキュベートし、比較のためにSECを行った。(C)分析用SECサンプルのピークから採取したサンプルのSDS PAGE分析。(D)TcdBドメインの組織図(上)と、トランスロケーションドメインに結合するsd5D(ヨード紫)と結合するTcdB構造(下、PDB:6OQ5)。(E)Vero細胞ベースの細胞毒性中和アッセイ。cSC5Dを含む標記分子による4pM TcdBの濃度依存的中和を、リガンドおよび非リガンドで示す。使用した全抗体の概略図を上に示す。
図S2
図をダウンロード
新しいタブで開く
図S2
図3および図S3に関連する: PA41抗体はTcdAとTcdBの両方を中和する。
(A)TcdA(50pM)とTcdB(4pM)のmIgA-PA41中和。(B)TcdAとTcdBのグルコシルトランスフェラーゼドメイン(GTD)の配列アラインメント;TcdBとPA41の接触残基をオレンジの四角で示す。
図S3
図をダウンロード
新しいタブで開く
図S3
図3および図S2に関連:二重特異性cS5DIgA-PA41の相乗的中和効果。
(A)cS5DIgA-PA41を試験するために特異的にVero細胞ベースの細胞毒性中和アッセイに使用した抗体の概略図。(B) TcdA (50pM)の濃度依存性中和。(C)TcdA(50pM)およびTcdB(4pM)の標記抗体による濃度依存的中和。(D)標記抗体によるTcdB(4pM)の濃度依存的中和。
図S4
図をダウンロード
新しいタブで開く
図S4
図4関連
(A)mSCBFPのSECクロマトグラム、吸光度スペクトル、蛍光スペクトル。 B)cSCCD5SLPとSLPの結合を示す分析SEC。(C)sdCD5SLPとSLPの結合を示す分析的SEC。(D)SCD5SLPFcα(リガンドcSCCD5SLP)とSLPの結合を示す分析的SEC。(E)分析SECサンプルのピークから集めたサンプルのSDS PAGE分析。
図S5
図をダウンロード
新しいタブで開く
図S5
図5関連:SLP結合部位が1つの抗体は架橋を促進しない
(A)C.difficileサンプルを標記抗体またはコントロールとインキュベートした8時間時点のフローサイトメトリーデータと、関連する共焦点顕微鏡画像。スケールバーは50μm。
図S6
図をダウンロード
新しいタブで開く
図S6
図5の関連図:増殖期のピークにおける異なるサンプルのC. difficileの凝集状態
(A) 成長中のC. difficile培養物に投与した、示した抗体またはコントロールの反復実験成長曲線。(B)100個以上/サンプルの手作業による計数によって決定された、指示された各サンプルにおける凝集細胞の割合(エラーバーは不均一なサンプル、および変数については統計的に当てはまらない)。(C)C.difficileに投与した抗体の代表的な共焦点顕微鏡像と明視野顕微鏡像。スケールバーは5μm。
図S7
図をダウンロード
新しいタブで開く
図S7
図6に関連:3時点におけるC. difficileの凝集状態。
(A)示したサンプルにおけるC. difficile培養の詳細を示す共焦点顕微鏡像。すべての画像のスケールバーは25μmである。
謝辞
この研究に関連した洞察に満ちた会話と示唆を与えてくれたStadtmueller研究室のメンバーに感謝する。Kritika Mehta、Kai Zhang博士、Elizabeth Roland博士、Collin Kieffer博士、UIUC Carl R. Woese Institute for genomic Biology Imaging Coreの顕微鏡サポートに感謝する。マイケル・ミラー研究室(UIUC)、特にC. difficile実験に実験室スペースとトレーニングを提供してくれたミラー博士と謝志帆に感謝する。Craig Ellermeier博士(アイオワ大学)にはC. difficileに関する洞察に満ちた議論を、Shannon Sirk博士にはC. difficile抗原(TXA1およびSLP)のプラスミドを提供していただいた。また、UIUCのRoy. J. Carver Biotechnology centerとCytometry and Microscopy to Omics (CMtO) FacilityにはフローサイトメトリーにBD LSR Fortessaを、UIUC School of Chemical Sciences High Throughput Screening facilityにはBiotek cytation 5プレートリーダーを提供していただいた。本研究は、NIH助成金1R01AI165570およびB.M.S.へのイリノイ大学スタートアップ資金の支援を受けた。
参考文献
1.↵ 清野秀雄およびD. W. PascualSilva-Sanchez, A., and Randall, T.D. (2020). 第2章 粘膜免疫・寛容の誘導と制御における粘膜免疫系(GALT, NALT, iBALT)の解剖学的独自性。Mucosal Vaccines (Second Edition), H. Kiyono and D. W. Pascual, eds. (Academic Press), pp.21-54. doi:10.1016/B978-0-12-811924-2.00002-X.CrossRefGoogle Scholar
2.↵Brandtzaeg, P., and Pabst, R. (2004). 粘膜に行こう:滑りやすい地面でのコミュニケーション。Doi:10.1016/j.it.2004.09.005.CrossRefPubMedWeb of ScienceGoogle Scholar
3.ȕBelkaid, Y., and Hand, T. (2014). 免疫と炎症における微生物叢の役割。Cell 157, 121-141. doi:10.1016/j.cell.2014.03.011.CrossRefPubMedWeb of ScienceGoogle Scholar
4.↵Brandtzaeg, P. (2013). 分泌型IgA:抗微生物防御のためのデザイン。Google Scholar
5.↵de Sousa-Pereira, P., and Woof, J.M. (2019). IgA: Structure, Function, and Developability. Antibodies 8, 57 doi:10.3390/antib8040057.CrossRefPubMedGoogle Scholar
6.↵Woof, J.M., and Russell, M.W. (2011). IgAの構造と機能の関係。Mucosal Immunol 4, 590-597. doi:10.1038/mi.2011.39.CrossRefPubMedWeb of ScienceGoogle Scholar
7.ȕDavis, S.K., Selva, K.J., Kent, S.J., and Chung, A.W. (2020). 感染症および癌における血清IgA Fcエフェクター機能。免疫細胞生物学 98, 276-286. doi:10.1111/imcb.12306.CrossRefGoogle Scholar
8.↵Monteiro, R.C. (2010). 炎症におけるIgAおよびIgA Fc受容体の役割。J Clin Immunol 30, 1-9. doi:10.1007/s10875-009-9338-0.CrossRefMedGoogle Scholar
9.↵Turula, H., and Wobus, C.E. (2018). The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 10, 237.doi:10.3390/v10050237.CrossRefPubMedGoogle Scholar
10.↵Kaetzel, C.S. (2005). 高分子免疫グロブリン受容体:粘膜表面における自然免疫応答と適応免疫応答の橋渡し。免疫学レビュー 206, 83-99. doi:10.1111/j.0105-2896.2005.00278.x.CrossRefPubMedWeb of ScienceGoogle Scholar
11.↵Mantis, N.J., Rol, N., and Corthésy, B. (2011). 分泌性IgAの腸管における免疫と粘膜恒常性における複雑な役割。Mucosal Immunol 4, 603-611. doi:10.1038/mi.2011.41.CrossRefPubMedWeb of ScienceGoogle Scholar
12.↵Royle, L., Roos, A., Harvey, D.J., Wormald, M.R., Van Gijlswijk-Janssen, D., Redwan, E.-R.M., Wilson, I.A., Daha, M.R., Dwek, R.A., and Rudd, P.M. (2003). 分泌型IgAのN型およびO型糖鎖は、自然免疫系と適応免疫系*の間のリンクを提供する。Journal of Biological Chemistry 278, 20140-20153. doi:10.1074/jbc.M301436200.Abstract/FREE Full TextGoogle Scholar
13.↵Pabst, O., and Slack, E. (2020). IgAと腸内細菌叢:特異的であることの重要性。Mucosal Immunol 13, 12-21. doi:10.1038/s41385-019-0227-4.CrossRefGoogle Scholar
14.↵Rochereau, N., Pavot, V., Verrier, B., Ensinas, A., Genin, C., Corthésy, B., and Paul, S. (2015). HIV抗原をM細胞に送達するためのワクチンキャリアとしての分泌性IgA。doi:10.1002/eji.201444816.CrossRefGoogle Scholar。
15.↵Mikulic, J., Bioley, G., and Corthésy, B. (2017). SIgA-Shigella Immune Complexes Interactes with Dectin-1 and SIGNR3 to Differentially Regulate Mouse Peyer's Patch and Mesenteric Lymph Node Dendritic Cell's Responsiveness. Journal of Molecular Biology 429, 2387-2400. doi:10.1016/j.jmb.2017.05.024.CrossRefGoogle Scholar.
16.↵Hoces, D., Arnoldini, M., Diard, M., Loverdo, C., and Slack, E. (2020). 流れるような環境で成長し、進化し、固着する:腸内細菌とIgAの相互作用を理解する。doi:10.1111/imm.13156.CrossRefGoogle Scholar
17.↵Hockenberry, A., Slack, E., and Stadtmueller, B.M. (2023). License to Clump: 分泌型IgAの構造-機能相関。この論文では、IgAの構造-機能相関のスケール間における関係を明らかにした。
18.↵Perrier, C., Sprenger, N., and Corthésy, B. (2006). 分泌成分上の糖鎖は、粘膜病原体に対する自然保護に関与する *. Journal of Biological Chemistry 281, 14280-14287. doi:10.1074/jbc.M512958200.Abstract/FREE Full TextGoogle Scholar
19.↵Maurer, M.A., Meyer, L., Bianchi, M., Turner, H.L., Le, N.P.L., Steck, M., Wyrzucki, A., Orlowski, V., Ward, A.B., Crispin, M., et al. ヒトIgAの糖鎖修飾は、インフルエンザAおよび他のシアル酸結合ウイルスを直接阻害する。Cell Reports 23, 90-99. doi:10.1016/j.celrep.2018.03.027.CrossRefGoogle Scholar
20.↵Finn, E., Andersson, F.L., and Madin-Warburton, M. (2021). クロストリジオイデス・ディフィシル感染症(CDI)の負担-一次性および再発性CDIの疫学に関するシステマティックレビュー。doi:10.1186/s12879-021-06147-y.CrossRefGoogle Scholar。
21.↵Feuerstadt, P., Nelson, W.W., Drozd, E.M., Dreyfus, J., Dahdal, D.N., Wong, A.C., Mohammadi, I., Teigland, C., and Amin, A. (2022). 高齢者におけるクロストリジオイデス・ディフィシル感染症の死亡率、医療利用、および費用。doi:10.1016/j.jamda.2022.01.075.CrossRefGoogle Scholar。
22.ȕVoth, D.E., and Ballard, J.D. (2005). クロストリジウム・ディフィシル毒素: 作用機序と疾患における役割。Doi:10.1128/CMR.18.2.247-263.2005.Abstract/FREE Full TextGoogle Scholar
23.ȕAl-Jashaami, L.S., and DuPont, H.L. (2016). クロストリジウム・ディフィシル感染の管理。Gastroenterol Hepatol (N Y) 12, 609-616.Google Scholar
24.ȕCastro-Córdova, P., Mora-Uribe, P., Reyes-Ramírez, R., Cofré-Araneda, G., Orozco-Aguilar, J., Brito-Silva, C., Mendoza-León, M.J., Kuehne, S.A., Minton, N.P., Pizarro-Guajardo, M., et al. (2021). 腸管上皮細胞への芽胞の侵入は、Clostridioides difficile感染の再発に寄与する。Nat Commun 12, 1140. doi:10.1038/s41467-021-21355-5.CrossRefPubMedGoogle Scholar
25.↵Song, J.H., and Kim, Y.S. (2019). 再発性クロストリジウム・ディフィシル感染症: 危険因子、治療、予防。Gut Liver 13, 16-24. doi:10.5009/gnl18071.CrossRefPubMedGoogle Scholar
26.↵Lee, Y., Lim, W.I., Bloom, C.I., Moore, S., Chung, E., and Marzella, N. (2017). クロストリジウム・ディフィシル感染症に対するベズロトクスマブ(ジンプラバ)。P T 42, 735-738.Google Scholar
27.↵Navalkele, B.D., and Chopra, T. (2018). Bezlotoxumab: An emerging monoclonal antibody therapy for prevention of recurrent Clostridium difficile infection. doi:10.2147/BTT.S127099.CrossRefGoogle Scholar。
28.↵Džunková, M., Moya, A., Vázquez-Castellanos, J.F., Artacho, A., Chen, X., Kelly, C., and D'Auria, G. (2016). 活性型および分泌型IgA被覆細菌分画は、クロストリジウム・ディフィシル感染におけるディスバイオシスを解明する。10.1128/msphere.00101-16.CrossRefGoogle Scholar
29.ȕStadtmueller, B.M., Huey-Tubman, K.E., López, C.J., Yang, Z., Hubbell, W.L., and Bjorkman, P.J. (2016). 分泌成分の構造と動態、および高分子免疫グロブリンとの相互作用。
30.ȕ Kumar Bharathkar, S., Parker, B.W., Malyutin, A.G., Haloi, N., Huey-Tubman, K.E., Tajkhorshid, E., and Stadtmueller, B.M. (2020). eLife 9, e56098. doi:10.7554/eLife.56098.CrossRefGoogle Scholar。
31.↵Kumar, N., Arthur, C.P., Ciferri, C., and Matsumoto, M.L. (2020). 分泌型免疫グロブリンAコアの構造。Science誌 367, 1008-1014. doi:10.1126/science.aaz5807.Abstract/FREE Full TextGoogle Scholar
32.↵Wang, Y., Wang, G., Li, Y., Zhu, Q., Shen, H., Gao, N., and Xiao, J. (2020). 分泌型免疫グロブリンAと肺炎球菌アドヘシンとの相互作用に関する構造学的知見。Cell Res 30, 602-609. doi:10.1038/s41422-020-0336-3.CrossRefGoogle Scholar
33.↵Hussack, G., Arbabi-Ghahroudi, M., van Faassen, H., Songer, J.G., Ng, K.K.-S., MacKenzie, R., and Tanha, J. (2011). 細胞受容体結合ドメインを標的とするシングルドメイン抗体によるクロストリジウム・ディフィシレ・トキシンAの中和。J Biol Chem 286, 8961-8976. doi:10.1074/jbc.M110.198754.Abstract/FREE Full TextGoogle Scholar
34.↵Murase, T., Eugenio, L., Schorr, M., Hussack, G., Tanha, J., Kitova, E.N., Klassen, J.S., and Ng, K.K.S. (2014). Clostridium difficile由来毒素AおよびBの受容体結合ドメインにおける抗体認識の構造的基盤。J Biol Chem 289, 2331-2343. doi:10.1074/jbc.M113.505917.Abstract/FREE Full TextGoogle Scholar
35.↵Orth, P., Xiao, L., Hernandez, L.D., Reichert, P., Sheth, P.R., Beaumont, M., Yang, X., Murgolo, N., Ermakov, G., DiNunzio, E., et al. クロストリジウム・ディフィシル毒素B中和抗体ベズロトクスマブの作用機序とエピトープがX線結晶構造解析により明らかになった。J Biol Chem 289, 18008-18021. doi:10.1074/jbc.M114.560748.Abstract/FREE Full TextGoogle Scholar
36.ȕAnosova, N.G., Cole, L.E., Li, L., Zhang, J., Brown, A.M., Mundle, S., Zhang, J., Ray, S., Ma, F., Garrone, P., et al. 3種の完全ヒトトキシンAおよびトキシンB特異的モノクローナル抗体の組み合わせは、ハムスターモデルにおいて、高病原性伝染性クロストリジウム・ディフィシル(Clostridium difficile)株のチャレンジから保護する。Clinical and Vaccine Immunology 22, 711-725. doi:10.1128/CVI.00763-14.Abstract/FREE Full TextGoogle Scholar
37.ȕChen, P., Lam, K., Liu, Z., Mindlin, F.A., Chen, B., Gutierrez, C.B., Huang, L., Zhang, Y., Hamza, T., Feng, H., et al. Nat Struct Mol Biol 26, 712-719. doi:10.1038/s41594-019-0268-0.CrossRefGoogle Scholar.
38.ȕKroh, H.K., Chandrasekaran, R., Zhang, Z., Rosenthal, K., Woods, R., Jin, X., Nyborg, A.C., Rainey, G.J., Warrener, P., Melnyk, R.A., et al. (2018). クロストリジウム・ディフィシル毒素TcdBの酵素カーゴの宿主細胞への送達を阻害する中和抗体。Journal of Biological Chemistry 293, 941-952. doi:10.1074/jbc.M117.813428.Abstract/FREE Full TextGoogle Scholar
39.↵Merrigan, M.M., Venugopal, A., Roxas, J.L., Anwar, F., Mallozzi, M.J., Roxas, B.A.P., Gerding, D.N., Viswanathan, V.K., and Vedantam, G. (2013). 表面層タンパク質A(SlpA)はクロストリジウム・ディフィシルの宿主細胞接着に大きく寄与する。PLOS ONE 8, e78404. doi:10.1371/journal.pone.0078404.CrossRefMedGoogle Scholar
40.↵Lanzoni-Mangutchi, P., Banerji, O., Wilson, J., Barwinska-Sendra, A., Kirk, J.A., Vaz, F., O'Beirne, S., Baslé, A., El Omari, K., Wagner, A., et al. C.difficileにおけるS層の構造とアセンブリー。Nat Commun 13, 970. doi:10.1038/s41467-022-28196-w.CrossRefGoogle Scholar
41.ȕCalabi, E., Calabi, F., Phillips, A.D., and Fairweather, N.F. (2002). クロストリジウム・ディフィシル(Clostridium difficile)表層タンパク質の消化管組織への結合。Infect Immun 70, 5770-5778. doi:10.1128/IAI.70.10.5770-5778.2002.Abstract/FREE Full TextGoogle Scholar
42.ȕLynch, M., Walsh, T.A., Marszalowska, I., Webb, A.E., MacAogain, M., Rogers, T.R., Windle, H., Kelleher, D., O'Connell, M.J., and Loscher, C.E. (2017). 病原性クロストリジウム・ディフィシル(Clostridium difficile)リボタイプ由来の表層タンパク質は、自然免疫反応に影響を及ぼす正の選択のシグネチャーを示す。BMC Evolutionary Biology 17, 90. doi:10.1186/s12862-017-0937-8.CrossRefGoogle Scholar
43.Mori, N., and Takahashi, T. (2018). クロストリジウム・ディフィシルにおける表層タンパク質の特徴と免疫学的役割.Ann Lab Med 38, 189-195. doi:10.3343/alm.2018.38.3.189.CrossRefGoogle Scholar
44.↵Calabi, E., Ward, S., Wren, B., Paxton, T., Panico, M., Morris, H., Dell, A., Dougan, G., and Fairweather, N. (2001). クロストリジウム・ディフィシル(Clostridium difficile)由来表面層タンパク質の分子学的特性解析。このような研究成果をもとに、クロストリジウム・ディフィシル(Clostridium difficile)由来の表面層タンパク質の分子学的特徴を明らかにした。
45.↵Drudy, D., Calabi, E., Kyne, L., Sougioultzis, S., Kelly, E., Fairweather, N., and Kelly, C.P. (2004). クロストリジウム・ディフィシル感染における表層タンパク質に対するヒト抗体反応。FEMS Immunology & Medical Microbiology 41, 237-242. doi:10.1016/j.femsim.2004.03.007.CrossRefPubMedGoogle Scholar
46.↵Kandalaft, H., Hussack, G., Aubry, A., van Faassen, H., Guan, Y., Arbabi-Ghahroudi, M., MacKenzie, R., Logan, S.M., and Tanha, J. (2015). シングルドメイン抗体による表面層タンパク質の標的化:クロストリジウム・ディフィシル関連疾患に対する治療アプローチの可能性。Appl Microbiol Biotechnol 99, 8549-8562. doi:10.1007/s00253-015-6594-1.CrossRefGoogle Scholar
47.↵Corthesy, B. (2013). 粘膜表面における分泌性IgAの多面的機能。Google Scholar
48.↵Gerhard, R., Frenzel, E., Goy, S., and Olling, A. (2013). Clostridium difficile TcdAおよび受容体結合ドメインを欠失した切断型TcdAの細胞内取り込み。クロストリジウム・ディフィシル(Clostridium difficile)TcdAおよび受容体結合ドメインを欠失した切断型TcdAの細胞内取り込み。
49.↵Manse, J.S., and Baldwin, M.R. (2015). クロストリジウム・ディフィシル(Clostridium difficile)毒素Bの結合と侵入は、複数のドメインによって媒介される。FEBS Letters 589, 3945-3951. doi:10.1016/j.febslet.2015.11.017.CrossRefGoogle Scholar
50.↵Warny, M., Pepin, J., Fang, A., Killgore, G., Thompson, A., Brazier, J., Frost, E., and McDonald, L.C. (2005). 北米および欧州における重症疾患の集団発生に関連するクロストリジウム・ディフィシルの新興株による毒素産生。CrossPubMedWeb of ScienceGoogle Scholar。
51.↵Richards, A.F., Baranova, D.E., Pizzuto, M.S., Jaconi, S., Willsey, G.G., Torres-Velez, F.J., Doering, J.E., Benigni, F., Corti, D., and Mantis, N.J. (2021). 組換えヒト分泌性IgAはサルモネラ・チフスムリウムの凝集を誘導し、腸関連リンパ組織への細菌の侵入を制限する。ACS Infect. Dis. 7, 1221-1235. doi:10.1021/acsinfecdis.0c00842.CrossRefGoogle Scholar
52.↵Moor, K., Diard, M., Sellin, M.E., Felmy, B., Wotzka, S.Y., Toska, A., Bakkeren, E., Arnoldini, M., Bansept, F., Co, A.D., et al. (2017). 高活性IgAは、増殖中の細菌を鎖でつなぐことで腸を保護する。Nature 544, 498-502. doi:10.1038/nature22058.CrossRefPubMedGoogle Scholar
53.↵ H. Sommermeyer and J. PiątekSommermeyer, H., and Piątek, J. (2021). 微生物学。クロストリジオイデス・ディフィシル:感染症、危険因子、予防と治療、H. Sommermeyer and J. Piątek, eds. (Cham: Springer International Publishing), pp.
54.↵Stadtmueller, B.M., and Kumar Bharathkar, S. (2023). キメラ分泌成分ポリペプチドとその用途.Google Scholar
55.↵Göritzer, K., Maresch, D., Altmann, F., Obinger, C., and Strasser, R. (2017). HEK293および植物産生ヒトIgAアイソタイプの部位特異的N-グリコシル化の探索。J. Proteome Res. 16, 2560- 2570. doi:10.1021/acs.jproteome.7b00121.CrossRefPubMedGoogle Scholar
56.↵Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. (1989). ポリメラーゼ連鎖反応を用いたオーバーラップ伸長による部位特異的突然変異誘発。遺伝子 77, 51-59. doi:10.1016/0378-1119(89)90358-2.CrossRefPubMedWeb of ScienceGoogle Scholar
57.Babcock, G.J., Broering, T.J., Hernandez, H.J., Mandell, R.B., Donahue, K., Boatright, N., Stack, A.M., Lowy, I., Graziano, R., Molrine, D., et al. トキシンAおよびBに対するヒトモノクローナル抗体は、ハムスターのクロストリジウム・ディフィシル誘発死亡を予防する。Infect Immun 74, 6339-6347. doi:10.1128/IAI.00982-06.Abstract/FREE Full TextGoogle Scholar
58.Laursen, N.S., Friesen, R.H.E., Zhu, X., Jongeneelen, M., Blokland, S., Vermond, J., Eijgen, A. van, Tang, C., Diepen, H. van, Oblomova, G., et al. インフルエンザヘマグルチニンに対するマルチドメイン抗体によるインフルエンザ感染に対する普遍的防御。doi:10.1126/science.aaq0620.Abstract/FREE Full TextGoogle Scholar
59.↵Sorg, J.A., and Dineen, S.S. (2009). クロストリジウム・ディフィシルの検査室での維持管理。doi:10.1002/9780471729259.mc09a01s12.CrossRefPubMedGoogle Scholar。
60.ȕEdwards, A.N., Suárez, J.M., and McBride, S.M. (2013). 嫌気環境におけるクロストリジウム・ディフィシルの培養と維持。J Vis Exp, 50787.doi:10.3791/50787.CrossRefGoogle Scholar
トップへ戻る
前へ 次へ
2023年11月12日掲載
PDFダウンロード
印刷/保存オプション
Eメール
共有する
引用ツール
COVID-19 SARS-CoV-2に関するmedRxivおよびbioRxivのプレプリント
テーマ領域
免疫学
対象分野
すべての記事
動物の行動と認知
生化学
生物工学
生物情報学
生物物理学
癌生物学
細胞生物学
臨床試験
発生生物学
生態学
疫学
進化生物学
遺伝学
ゲノミクス
免疫学
微生物学
分子生物学
神経科学
古生物学
病理学
薬理学と毒性学
生理学
植物生物学
科学コミュニケーションと教育
合成生物学
システム生物学
動物学
bioRxiv の臨床研究パイロットプロジェクトが終了し、健康科学専用サーバー medRxiv (submit.medrxiv.org)が開設されたことに伴い、Clinical Trials と Epidemiology のサブジェクトカテゴリーは新規投稿を締め切りました。臨床試験の結果を報告する新規論文は、medRxivへの投稿が必須となりました。ほとんどの疫学論文もmedRxivに投稿されるべきですが、もし論文に健康に関する情報が含まれていない場合、著者は他のbioRxivサブジェクトカテゴリー(例えば、遺伝学や微生物学)に投稿することもできます。
コンテキストと評価 x
0 0 0 1 0 0 11
この記事が気に入ったらサポートをしてみませんか?