
マイクロバイオームデータにおけるdifferential abundance法の検討: ベンチマークスタディ

研究論文
マイクロバイオームデータにおけるdifferential abundance法の検討: ベンチマークスタディ
https://journals.plos.org/ploscompbiol/article/figures?id=10.1371/journal.pcbi.1010467
マルコ・カッペラート、ジャコモ・バルッツォ、バルバラ・ディ・カミッロ
バージョン2 バージョン1 無修正プルーフ
本論文は、PLOS Computational Biology Benchmarkingの論文です。
概要
効率的で費用対効果の高いハイスループットDNAシーケンサー技術の開発により、複雑な微生物システムを研究する可能性が高まっています。近年では、異なる生態系ニッチを特徴づける微生物の研究に大きな関心が集まっています。存在量差分解析は、2つのクラスの被験者またはサンプル間の各分類群の存在量の違いを見つけることを目的とし、各比較に有意値を割り当てる。バイオインフォマティクスの手法は、スパース性、サンプル間のシーケンスの深さの違い、組成の違いといったマイクロバイオームデータの課題を考慮して開発されたものである。存在量の差分解析は、健康から環境まで、さまざまな分野で重要な結論を導いてきた。しかし、既知の生物学的真実がないため、得られた結果を検証することは困難である。本研究では、微生物シーケンスのカウントデータシミュレータであるmetaSPARSimを利用して、実験グループ間の存在量の差の特徴を持つデータをシミュレートします。シミュレーションシナリオと評価指標の両方の信頼性に配慮し、共通のベンチマークで最近開発された方法と確立された方法の完全な比較を行う。性能の概要には、多数のシナリオの調査が含まれ、サンプルサイズ、有意差のある特徴の割合、シーケンスの深さ、特徴のばらつき、正規化のアプローチ、生態学的ニッチなどの主要な共変量が手法の結果に与える影響を調査しています。主な結果は、データセットとサンプルサイズに依存するようですが、メソッドは高いサンプルサイズにおいて、タイプIエラーと一般的に、偽発見率をうまくコントロールできることがわかりました。
著者概要
微生物叢とは、生態学的環境またはニッチを特徴づける微生物の集合体である。いくつかの研究により、微生物叢が宿主生物や生態系の健康やバランスに影響を与える様々な生物学的メカニズムに関与していることが示されています。バイオインフォマティクスの計算手法の発展とともに、ますます効率的になった配列決定技術により、新たな発見や洞察が可能になりました。このような状況の中で、最も興味深い解析のひとつは、2つのグループを分析したときに、有意に異なる存在量を示す微生物の同定である。多くの計算手法が開発されているが、どの計算手法が最も優れた性能を持つかはまだ不明である。そこで我々は、マイクロバイオームデータのシミュレーターを利用してシミュレーションフレームワークを構築し、存在量差分解析の既知のツールの広範なベンチマークを実施することができました。私たちの研究は、解析者がツールを選択する際の指針となるだけでなく、堅牢で信頼性の高い公正なシミュレーションフレームワークに向けた第一歩となる。
引用元 Cappellato M, Baruzzo G, Di Camillo B (2022) Investigating differential abundance methods in microbiome data: ベンチマークスタディ。PLoS Comput Biol 18(9): e1010467. doi:10.1371/journal.pcbi.1010467
編集者 中国・復旦大学 ルイス・ペドロ・コエルホ氏
受理された: 2021年12月23日、受理された: 2022年8月3日、発行: 2022年9月8日発行
著作権:©2022 Cappellato et al. これは、原著者と出典をクレジットすることを条件に、あらゆる媒体での無制限の使用、配布、複製を許可するクリエイティブ・コモンズ表示ライセンスの条件の下で配布されるオープンアクセス記事です。
データの利用可能性 この出版物をサポートするために書かれたすべてのコードは、https://gitlab.com/sysbiobig/metabenchda で一般に公開されています。シミュレーションの入力ファイルと生成されたデータは、https://doi.org/10.5281/zenodo.5799193 から入手できます。
資金提供 本研究は,パドバ大学情報工学科のSEEDプロジェクト「tRajectoriEs of baCtErial NeTwoRks from hEalthy to disease state and back(RECENTRE)」(Grant nr. DI_C_BIRD2020_01(BDC))から支援を受けています.G.B.は、PON 'Ricerca e Innovazione' 2014-2020によって設立されました。資金提供者は、研究デザイン、データ収集と分析、発表の決定、原稿の作成に関与していない。
競合する利益 著者らは、競合する利益が存在しないことを宣言している。
はじめに
次世代シーケンサー(NGS)は、多くの疾患における微生物集団の役割について視野を広げており[1-4]、サンプルグループ間の表現型の違いの原因となる特定の分類群を特定することができるとして、差分存在量(DA)解析[5]にますます注目が集まっています。しかし、最近Wallen [6]が2つの大規模なパーキンソン病腸管16Sデータセットに適用して指摘したように、結果は適用したDA手法に大きく依存する。さらに、マイクロバイオームシーケンスのカウントデータの前処理は結果に強く影響し、その後の統計解析で誤った解釈をしないためには、適切なデータ処理が基本になります [7] 。第一に、カウント表は非常にまばらであり、すなわちヌル値の割合が高い。第二に、存在量に関する情報は、サンプルに存在する特定の分類群の要素の総数を反映するものではなく、その一部、すなわちサンプルのシーケンス深度に対する相対的なものであることです。実際、データは相対的な情報を伝え、組成的であり、Simplexと呼ばれる部分空間に存在する[12]。最後に、配列決定プロセスから得られる配列決定深度は、一般的にサンプル間で異なる。
基本的に、2つの異なる集団における個々の分類群のカウントをテストする場合、対処すべき2つの大きな問題がある。Linら[13]が提案したおもちゃの例に従って、同じ配列深度4でサンプリングされた2つの分類群t1、t2からなる2つの生態系A = [t1A = 4, t2A = 4] とB = [t1B = 6, t2B = 6] を考えてみましょう。 観察結果は、A' = [t1A' = 2, t2A' = 2] と B' = [t1B' = 2, t2B' = 2] です。観測された集団A'とB'を比較すると、2つの分類群のどちらも差分的に豊富ではないと誤って結論づけることができます。さらなる例として、t1の個体数がAに対して大幅に増加する第3の生態系C = [t1C = 12, t2C = 4]を考えてみましょう。この場合もシーケンスの深さが一定であると仮定すると、観測される個体数はC' = [t1C' = 3, t2C' = 1] です。この場合、A'とC'を比較すると、両分類群とも差分的に豊富であるが、Cではt1のみがより豊富であるという結論になりかねない。
上記の例では、差次的存在度の高い分類群の定義には、対象となる生態系の「真の絶対的存在度」が含まれています。しかし、研究者は、サンプル内の他の集団に対する存在量の変化に興味がある場合もあります。この場合、「真の相対的存在量」と呼ばれるt2に対するt1の割合が2つの集団で変化しないので、集団AとBの分類群は差次的存在量とはみなされません。この2つのアプローチのどちらを選択するかは、生物学的な要求に依存し[14,15]、研究者は絶対的な存在量の変化か相対的な存在量の変化に焦点を当てるようになる。
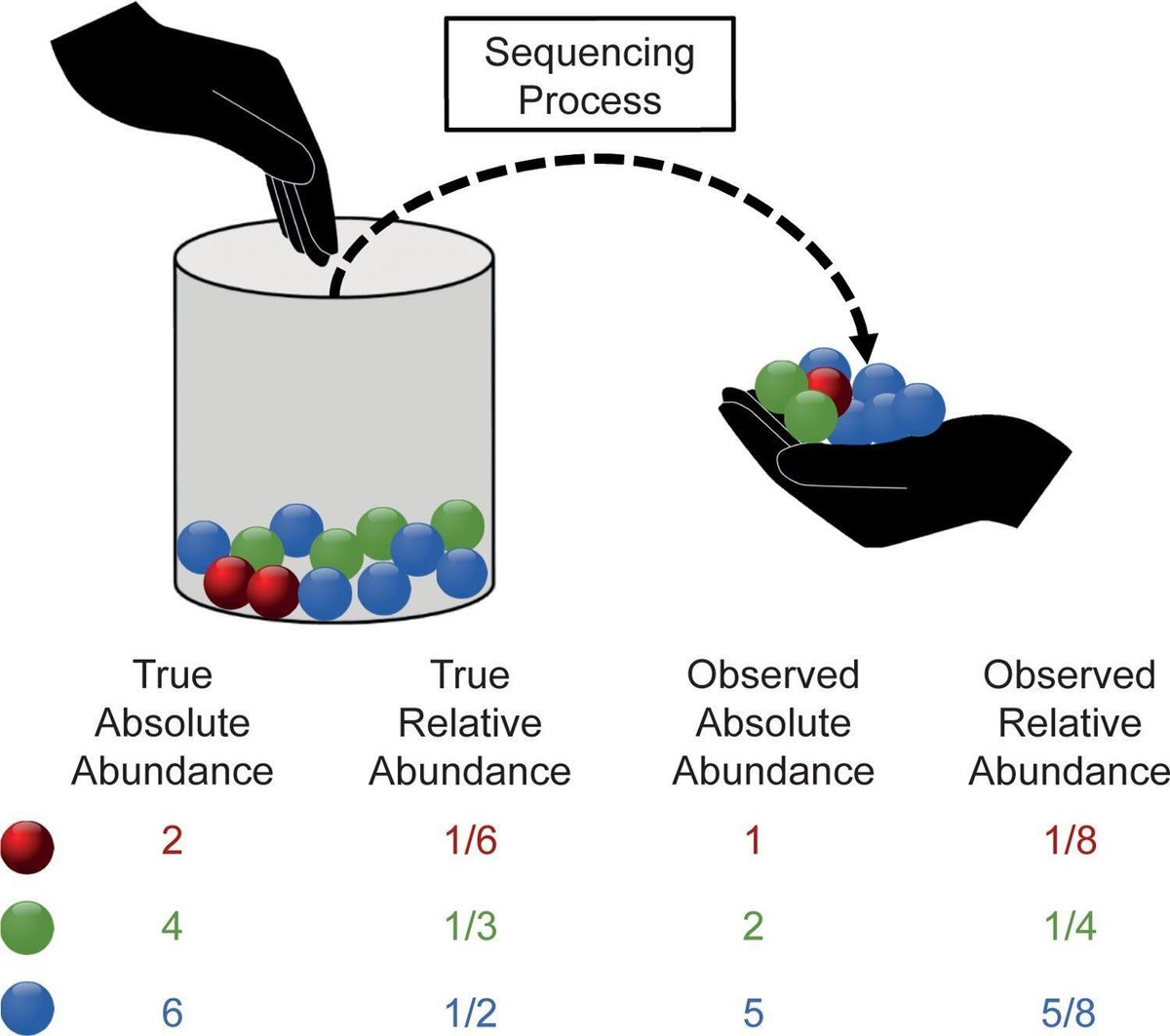
Linら[13]が以前指摘したように、多くの著者が相対的存在量と絶対的存在量という用語を使い分けているため、手法だけでなく他のベンチマーク研究の結果の解釈も難しくなっている。したがって、明示的な表記が望まれる。上記の例で使用されたすべての用語を図1にまとめた。特に、「真の絶対量」は分析した検体中の細菌の数を意味し、「真の相対量」は割合、すなわち総数に対する細菌の割合を意味します。これらの量がシーケンシングプロセスの出力を指す場合、「観察された」という用語を使用します。すなわち、「観察された絶対量」はシーケンシングプロセスによって測定されたカウント数であり、「観察された相対量」は総カウント数に対する対応する比率を指します。
拡大する
図1. 表記の概要。
壷は実際の微生物集団を表し、異なる細菌が異なる色の球体で表現されている。シーケンシングプロセスは、限られた抽出回数(すなわちシーケンシング深度)で置換なしのサンプリングによって記述することができます。
doi:10.1371/journal.pcbi.1010467.g001
詳細 "
この研究では、共通のベンチマークを用いた異なるDA手法の比較を提案します。16SメタゲノミクスデータのDA解析のために最近開発され、確立された手法と、この種の調査に最初に使用されたRNAシーケンスの分野から適応された手法を比較対象とする。
また、DA解析のために開発された手法のほとんどがこの分野であるため、単変量法、すなわち、被験者のグループ間で各分類群の存在量を個別に比較するツールに焦点を当てました[16]。我々の貢献は、既に発表されている他の比較研究[11,13,15,17-20]に関して、より多くのメトリクス、すなわち偽陽性率、偽発見率、Recall、精密-再現曲線、PR曲線下の部分面積、計算負荷で方法の性能を評価していることにある(S1ファイルの表A参照)。さらに、サンプルサイズ、DA分類群の割合、シーケンス深度、Fold Change、分類群の変動性、低存在DA分類群を回避/許可するための閾値の使用、ゼロエントリーを扱うための異なるアプローチ、正規化、異なる生態系ニッチなどの他のアプローチではまだ調査されていないシナリオや共変数を考慮します(S1ファイルの表Bを参照)。さらに、信頼性が高く、簡単に拡張できるシミュレーションのフレームワークを提案する。
文献では、シーケンスデータの生成にいくつかのモデルが使用されているが[11,13,15,17-20]、マイクロバイオームデータに最も適合するモデルに関する文献上の合意はまだ得られていない[19,21,22]。さらに、比較における潜在的なバイアスを避けるために、データのシミュレーションとDA分類群の検出に使用する生成モデルは同じであってはならない。
そこで、ベンチマーク研究で使用したデータをシミュレートするために、16Sデータの組成的性質を再構築する優れた能力を示しており、性能に対するパラメータの影響を調べることができ、評価に関与するツールがこのモデルを前提としていないことから、metaSPARSim [23] によって導入されたガンマ-多変量超幾何生成モデルを使用しました。特に、metaSPARSim [23]は、実際のデータセットから学習したパラメータから出発して、DA特徴を含む2つの実験グループをシミュレートするために使用されています。文献[11,15,17-20]で報告されているように、DA分類群の定義は、平均的な特徴の存在量のフォールドチェンジ(FC)に基づいている。特に、Khomichら[20]で行われたように、より現実的なシナリオに似せるために、FC値をあらかじめ定義された間隔で変化させます。さらに、我々のアプローチでは、FCアプローチで作成されたDA特徴の後にも、実データから学習した平均分散関係を維持することで、すでに報告されているように、非現実的な存在量分布を作成するリスクを防止しています[23]。
DA解析のための文献で使用されている統計モデルは、「絶対量」の変化と「相対量」の変化のどちらかを検証するために設計されている[13]。両方のシナリオを考慮するために、我々はシーケンスデータの構成性、すなわち、ある分類群の存在量の変化が他の分類群の観察された存在量に影響を与えるという事実を考慮してシミュレーションのフレームワークを開発しました[7-11]。特に、過剰発現する分類群と過小発現する分類群のバランスをとり、シミュレーションしたDA分類群が真の絶対量と真の相対量の両方で異なる発現量となるシナリオを作成しました。その結果、本シミュレーションは、絶対量に差のある分類群を検定する方法と相対量に差のある分類群を検定する方法の両方を評価するために使用することができる。
結果の再現性を確保するため、シミュレーションデータと評価スクリプトは、https://gitlab.com/sysbiobig/metabenchda で利用可能な metaBenchDA R パッケージに含まれました。開発されたRパッケージとテストされたDAメソッドを含むDockerコンテナイメージは、同じリンク先で入手できます。さらに、すべてのシミュレーションデータセット、メソッドの出力、評価メトリクスの結果を含むフォルダは、Zenodoのhttps://doi.org/10.5281/zenodo.5799193 で公開されています。
材料と方法
ディファレンシャルアバンダンスメソッドを評価
本研究では、マイクロバイオーム解析用に特別に開発された確立された最新のDA手法(ALDEx2 [24], eBay [25], ANCOM [26], ANCOM-BC [27], corncob [28], MaAsLin2 [29], metagenomeSeq [30] )およびRNA-seqデータの差分発現解析用に開発されたが16Sシーケンスデータ解析にも日常的に使用されている2つの手法( edgeR [31] and DESeq2 [32] )を対象にしています。
表1は、本ベンチマークスタディで検討した手法をまとめたもので、利用可能なソフトウェア、基本的なアプローチ、ツールで実行する前処理、採用したDA分類群の定義、すなわち各分類群の平均真の絶対量または相対量の有意差の調査を目的としている場合についての情報を示しています。
拡大する
表1. 本ベンチマークスタディに含まれる手法の概要をアルファベット順に示す。
doi:10.1371/journal.pcbi.1010467.t001
詳細 "
DESeq2、edgeR、metagenomeSeqは、FCを推定することを目的に、異なる統計モデルを用いて各分類群の観測絶対量をモデル化しています。ANCOMとANCOM-BCは、真の絶対量を目標に明示的に設計されています。表1に示した他のすべての方法は、(ALDEx2およびeBayの幾何平均を基準とした)差分相対存在量をターゲットとするツールに分類されます。手法のより詳細な説明は、S1ファイルのセクション2に記載されている。
ベースライン法として、ノンパラメトリックで、比較されるランダム変数の分布について仮定しないウィルコクソン順位和検定を選択しました。この検定は、平均ライブラリサイズ(AVG)によるデータのスケーリングと中心対数比(clr)変換という2つのベースラインデータ変換と組み合わせて使用する。
いくつかの例外を除いて、すべてのメソッドをデフォルトの設定で実行しました。metagenomeSeqの場合、Bioconductorパッケージのデフォルトを使用する代わりに、パッケージのヴィネットで元々提案され、以前のベンチマーク研究[19]にも従った手順で、CSS(Cumulative Sum Scaling)正規化 [30] が使用されました。DESeq2の場合、メタゲノムデータで示唆されているように、非NULL値のみを用いて幾何平均を計算した[33]。
すべてのメソッドで、False Discovery Rateの標準的な閾値5%を使用し、Benjamini-Hochberg(BH)手順で多重検定の補正を行った。このアプローチは、ANCOM-BCを除けば、検討中のすべてのツールのデフォルトであり、著者らはより保守的なHolm手順[34]の使用を推奨しています。
16Sおよびメタゲノミクスデータは、DNA抽出、PCR、および配列決定ステップが他の分類群よりも特定の分類群を優先的に測定および増幅するため、実験ワークフロー全体を通して実験バイアスの影響を受け、分類群およびプロトコル固有のバイアスで測定値を歪める[35, 36]。通常、この側面は、主に同じ分類群や実験内のサンプル間の比較を目的とした正規化手法によって部分的に対処されるだけです。しかし、異なる分類群間の比較や異なるプロトコルのデータを用いた比較は、この問題に対処しなければ不可能である。この点は我々の研究の範囲外であるが、表1では、異なるツールが分類群バイアスに対処できるかどうかを指定した列を報告している(詳細はS1ファイルのセクション2を参照)。
豊富な分類群による16Sカウントデータのシミュレート
16Sカウントデータのモデリング
このベンチマークスタディでは、16S微生物カウントデータのシミュレーションにmetaSPARSim [23]が想定する生成モデルを使用します。その理由は、(1)異なる実験シナリオで広範囲にテストされている、(2)評価中のツールによるDA機能の検出に使用されない生成モデルに基づいており、評価におけるバイアスを回避できる、(3)マイクロバイオームのシーケンスデータの基盤となる技術および生体特性に関わるパラメータに対応できる、などです。
metaSPARSim [23]が想定するカウント生成モデルは、2つのステップで構成されています。第一段階として、同じ実験グループに属する生物学的複製物間の絶対量レベルXijをモデル化するためにガンマ分布が使用されます:
(1)
以下、i は分類群、j は試料、k は実験群を示す。式1において、k群における分類群iの存在量レベルの生物学的変動を記述する分散値、k群のi番目の特徴量の平均存在量である。
第二段階として、多変量超幾何分布(MHG)を用いて、シーケンシングプロセスで発生する技術的なばらつきをモデル化する:
(2)
ここで、Yjはサンプルjの観察された分類学的プロファイルを表すベクトル、Ljはサンプルjのシーケンス深度(リードの総数)、Xjはjの絶対存在レベルのベクトルである。
上記の生成モデルを用いたシミュレータは、グランドトゥルース(ガンマ分布サンプリングの出力)とカウントテーブル(MHG分布サンプリングの出力)の2つのマトリックスを返します。
DAタクサのシミュレーション
2つの実験グループにまたがる存在量行列をシミュレーションするためには、metaSPARSimにベクトル(μ、φ、L)の異なるタプルを与える必要があります;すなわち、グループAのサンプルには(μA、φA、LA)、グループBのサンプルには(μB、φB、LB)です。条件A(μA、φA、LA)が与えられたとき、2つのグループ間でDA特徴を生成できるパラメータを設定することで、新しい実験グループB(μB、φB、LB)のタプルを作成する。
まず、条件AのシミュレーションパラメータをmetaSPARSimの組み込み関数(S1ファイルの3節参照)を用いて、または適応して推定します。グループAのパラメータは、異なる生物学的および技術的シナリオを表す3つの異なる実データセット[37-40]から推定する(S1ファイルの表2およびセクション4)。以下では,各データセットに,パラメータを推定した参照条件の名前を付ける(表2の5列目).条件A(μA、φA、LA)のシミュレーションパラメータを推定した後、シミュレーションプロセスの次のステップは、条件B(μB、φB、LB)の新しいパラメータを作成することです。このとき、条件Bには条件Aとは異なる豊富な分類群が含まれます。
拡大する
表2. 条件Aのパラメータの推定に使用したデータセットの概要。
doi:10.1371/journal.pcbi.1010467.t002
詳細 "
これまでの比較[11,17,19,20]と同様に、DA分類群の導入には、平均存在量のフォールドチェンジ(FC)に基づくアプローチが用いられている。より詳細には、平均絶対量にフォールドチェンジFCiを適用し、以下の場合に特徴iをDAと定義する:
(3)
ここで、FCi∈[f1,f2]の場合は、条件Bにおける特徴量iの存在量の増加を得、一方、存在量の減少を得る。重要なのは、この値が条件Aで観測された値の範囲内で、次のように制約されることである:
(4)
この研究では、f1 = 2とf2 = 5を設定し、先行研究[11,15,17-19]でテストした一定のFC値と一致するFC値の範囲を得た(S1ファイルの表Bを参照)。以前のベンチマーク研究[13,17,19,20]に従い、モデルのカウント行列出力(すなわちY)ではなく、生成モデルのパラメータ(すなわちμ)に作用して、差分的に豊富な分類群を定義することにしました。このように、技術的なノイズを加える前に、分類群の存在量の差をシミュレートすることが可能です。
DA特徴の望ましい割合(P)から出発して、どの特徴をDAとしてシミュレートするかは、その分布を考慮して選択される。 微生物カウントデータの存在量分布は、存在量の多いものに比べて存在量の少ないOTU/ASV値が多く、左寄りの傾向が強い(図2)。したがって、原理的にはDA特徴を模擬する候補をランダムにサンプリングできたとしても、実際には、低存在のDA特徴候補(図2の黒い区間)を多く選ぶことになり、配列決定プロセスによる制約から、カウント行列Yの一方または両方のグループでゼロになる可能性があります。さらに、低アバンダンス領域では、シーケンシングによってもたらされるノイズが高く、多くのDA特徴がシーケンシングプロセスによって不明瞭になる可能性がある。実際、多くの研究では、分析を行う前に低存在のOTU/ASV値をフィルタリングしています。同様に、DA法で検出するのが非常に困難な模擬分類群のラベリングを避けるため、グループAで平均して固定閾値θより高い存在度を持つものの中から、DA機能の候補をサンプリングしました。
拡大する
図2. 歯データセットの平均存在量分布(対数スケール)。
点線は、DAの特徴をサンプリングするための存在量レベルの限界値を示す:低(黒)、中(青)、高(赤)。
doi:10.1371/journal.pcbi.1010467.g002
詳細 "
θとΩの閾値で低存在域、中存在域、高存在域を定義することで、中存在域や高存在域のOTUの中から条件Aのi番目のOTUをDA特徴候補としてランダムに選択した場合に、異なるシナリオが発生する:
もし、 , がグループ A の中程度の存在量にある場合(図 2 の青字)。FCiは等確率で、つまり条件Aと比較して条件Bで過剰または過小なOTUをシミュレートする確率が等しくなるように選択される。
もしそうなら、A群(図2の赤色)の存在量が高い範囲にあることになる。したがって、FCiは、結果が.よりも低くなるように選択される、すなわち.である。
中域のDA候補特徴量(図2の青色)を考えると、条件BでDA候補特徴量がup表現された場合、式4によりその最大値はMに拘束される。そこで、条件Bでは、条件Aで観測可能な限界値を超えるような特徴を避けるように設定する(つまり、実験データを用いて、もっともらしい存在量の範囲を設定する)。同じ理由で、FCiの上限値は.FCiとする。一方,DAフィーチャーについては,条件Bの最小値を. θ=1-f2とし、DAフィーチャーの存在量の最小値が一様な平均値を持つようにする。最後に、平均アバンダンスベクトルの左寄りの分布から、高アバンダンス区間(図2の赤色)でのサンプリング確率が低いことに注意する必要がある。その結果、高存在域はDAフィーチャーが過小にしかシミュレートされない唯一のものであるため、全体として過不足シナリオはかなりバランスが取れている。なお、条件Bの平均絶対量に範囲を設定したのは、もっともらしい範囲のデータを生成するためである。しかし、このことは、結果として得られるカウントテーブルにおいて、2つの条件の強度の最大値と最小値が全く同じであることを意味するものではない。実際、μはガンマ分布の入力として提供され、生物学的ノイズ(式1)と技術的ノイズ(式2)を適用した後、2つの条件は、実際の実験条件で観察される値の範囲にほぼ収まるものの、平均最大観察存在量が異なると考えるのが妥当であろう。
追加調査として、θ = 0に設定し、低存在のDA特徴が存在する場合の手法のパフォーマンスを調査しました。この場合、DA分類群は全範囲でサンプリングされ、再び、上述の手順により、2つのシミュレーション条件間の過剰/過小な存在感のバランスが確保されます。
DA分類群: 絶対量と相対量の違い。
Weissら[11]で最初に指摘されたように、絶対量の変化としてDA特徴を導入すると、相対量の変化として他の全ての特徴に影響を与える可能性があることに注意すべきである。したがって、相対量を対象とした異なる手法では、意図的にDAとしてシミュレートされていない多くの特徴を関連するものとして正しく識別することができるかもしれない。
先行研究[11,17,19]では、FC適用後の非DA特徴のバランスについて、様々な方法で調査されている。Weissら[11]は、FCが2つの実験グループを交互に掛け合わせることで、シミュレートされたすべてのDA特徴の複製に基づくアプローチを提案した。この方法によって、著者らは、絶対量と相対量の両方においてDAである特徴を持つ、バランスのとれたシミュレーションを作成した。一方、Hawinkelら[17]では、「補償」と呼ばれるアプローチが導入されている。つまり、選択されたDAフィーチャーの一部(真の相対存在量の和がa)の平均真の相対存在量にFCを乗じ、残りのフィーチャー(真の相対存在量の和がb)には.を乗じる。このように、DA特徴を導入しても、同じように存在する分類群の平均相対存在量は変化しない。残念ながら、この方法はFC値が一定である場合にのみ可能であり、現実に近いシナリオとは言い難い。
そこで、ベンチマーク研究では、過剰・不足の分類群をバランスよく配置し、真の絶対量と真の相対量の両方で、模擬DA分類群の存在量が異なるシナリオを作成しました(図1)。この性質は、S1 FileのFig.1-3に示すように、事後的に検証されたので、以下に説明する。
絶対量が異なる特徴をシミュレートするためのFold Changeアプローチ(式3)に従い、同じ定義を使って相対量が異なる分類群(またはProportionally Differentially Abundant, PDA)を定義します。以下の場合、タクソンiはPDAである:
(5)
ここで、実験グループA:の真の相対存在量の平均値である。
S1ファイルの図1-3では、MVAプロット、すなわち、すべてのシミュレーションシナリオにおける2つのグループ(すなわち、および)の相対存在量の対数スケール(ベース2)の平均値に対するFCが、1つのランについて示されている。DA特徴(赤色)は、式3に基づいて絶対DAと定義されたものである。S1 FileのFig.1-3から明らかなように、絶対的なDAの特徴は、ほとんどのケースで2以上のフォールドチェンジを持つPDAでもあり、常にDAではない特徴(黒い点)とよく区別される。
上記の手順でμBパラメータを定義した後、実際のデータで観察される平均変動傾向に類似するように対応するφBパラメータを設定した。実際、OTUの存在量変動はOTUの平均存在量と連動しており、DA OTUに設定すると非現実的なデータになってしまう。そこで、DA OTUsのパラメータは、(μA, φA)で観察される傾向に類似するように設定される。簡単に説明すると、グループBで平均値 , を持つDA特徴をシミュレートする場合、グループAの平均存在量aに対応する観測値の線形補間に基づいて、ofの値が割り当てられる。
最後に、配列深度LBのベクトルは、観察された配列深度値の条件Aをランダムにサンプリングして得られる(S1ファイルのセクション3)。
評価シナリオ
まず、metaSPARSimを使用して、同じパラメータ値(μ、φ)を持つ2つのグループ、すなわち、DA機能を持たないシナリオをシミュレーションする。この設定では、異なるサンプルサイズ([10,25,50,100]のSS)、すなわち各実験グループのサンプル/被験者の数を、AグループとBグループで同じSSでテストします。
次に、前述したようにB群でDA分類群のシミュレーションを行い、A群の平均値に対して閾値θ=1-f2を設定し、低存在・高ノイズの文脈でDA特徴をシミュレーションすることを回避しました。この景観では、[13]で行われたのと同様に5%-15%-20%に等しいPを挿入して、手法の性能をテストした(S1ファイルの表Bを参照)。ここでも、サンプルサイズは先に述べたシミュレーションシナリオと同じに設定されている。
DA特徴を用いた上述のシミュレーションシナリオに加えて、平均シーケンス深度が最大のデータセット、すなわち非IBDを、Lオリジナルベクタを半分にすることと2倍にすることの両方によってシミュレーションした。シミュレーションの段階でランダムな種を設定し、これら2つのバリエーションにおける各シミュレーションで全体の手順が同じになるようにしました。その結果、これらの新バリアントにおける性能の違いは、シーケンスの深さにのみ依存します。さらに、マイクロバイオーム解析では、条件Aと条件Bで平均シーケンシング深度が異なる可能性があります。そこで、条件BのLを2倍にすることで、不均衡なシーケンス深度のシナリオを作り、IBD以外のシナリオもシミュレートしてツール性能を検証しました。
さらに、パラメータφ、すなわち絶対量の分散(式1)をシミュレータの入力とみなし、その効果を評価した(他のパラメータは前述のように設定した)。前回と同様に、観測された差異がパラメータφのみによるものであることを保証するために、ランダムシードを設定した。最終的なベンチマークとして、θ=0とし、平均存在量がかなり低くても、差分的に存在する分類群が存在するようにしました。このように、非常に難しいが現実的なシナリオであっても、手法の性能を評価することができた。
デフォルトの正規化を変更することで手法の性能が変わるかどうかを調べるために、本研究で検討したどのDA手法でも使用されていない、マイクロバイオームデータ用に特別に開発された正規化(GMPR [41])で手法をテストすることにした。正規化やデータ変換は使用するモデルの不可欠な部分であるため,ALDEx2,ANCOM,ANCOM-BC,corncob,eBay などのメソッドは,入力として生データを必要とすることを強調しておく(S1 File のセクション 2 参照).しかし、今回のシミュレーションでは、比較のために、正規化されたデータをすべての手法の入力として与えた。
最後に、上記のすべてのシナリオでツールのベンチマークに使用したシミュレーションデータセットは、人間に関連する環境に由来するものであることに留意すべきである(表2参照)。そこで、生態系ニッチを変えた場合にツールの性能が異なるかどうかを検証するため、ヒトに関連しないデータセットとして、ニワトリの腸内細菌叢(「AnimalGut」データセット)と土壌コミュニティ(「Soil」データセット)を用いて追加調査を行った(セクション4およびS1ファイルの表C参照)。
すべてのデータセットで調査した共変量のすべての組み合わせについて、上記のすべてのシミュレーションシナリオにおいて、50回のシミュレーションを実施した。
評価スコア
DA の特徴を正のクラスとし、同様に豊富な分類群を負のクラスとすると、各分類群の分類結果は、真陽性(TP、すなわち正しい陽性割り当て)、真陰性(TN、すなわち正しい陰性割り当て)、偽陽性(FP、すなわち誤った陽性割り当て)、偽陰性(FN、すなわち誤った陰性割り当て)になります。
DA分類群が存在しないシナリオでは、FPとグラウンドトゥルースの否定的な総数(FPとTNの合計)の比として定義される非調整p値で偽陽性率(FPR)を評価しました。このシナリオでは,メソッドによってDAと識別されたすべての特徴がFPであるため,FPの値は5%程度と予想される.
5%、15%、20%のDA特徴を持つシナリオでは、メソッドのパフォーマンスをテストするために、リコール、精度、偽発見率(FDR)、FPRも使用される。Recallは、正しく識別されたポジティブイベントの割合、Precisionは、ポジティブクラス全体に対するTPの割合として定義され、Hawinkelら[17]が示唆するように、特異性の代わりにこれを使用します。FDRは、FPの割合として定義される(したがって、FDR = 1-precision)。各手法で採用されている補正手順は、FDRの期待値を制御することを目的としている。最後に、このシナリオでは、先に定義したFPRを調整したp値で計算する。
さらに、精度と想起の間のトレードオフを示すために精度-想起(PR-)曲線、DAであることの証拠に基づいて特徴をランク付けする手法の全体的な能力を定量化するためにその下の面積(AUPR)が使用されている。我々は先行研究[18,19]の考えに従い、ランキング上位の特徴のみを含むAUPRの部分にも着目している。したがって、精度値の1~0.9の間の部分AUPR(pAUPR)を計算し、望ましい精度値の狭い範囲のみを考慮する。さらに、精度-想起性能を要約するために、各検討方法とシミュレーションパラメータの構成について、50回のシミュレーションにおけるi番目の精度値とi番目の想起値の平均として平均PRカーブを定義する。
最後に、各手法の計算時間を調査した。すべての方法は、Intel Core i7-7700 CPU @ 3.60GHz (4コア、8スレッド)と16GBラムを備えたシステムで実行されています。実行時間はRの基本関数 "Sys.time "を用いて,パラメータPについて考慮した3つの異なる値で得られたシミュレーションを平均して計算した(S1ファイルのセクション6).実際には,研究者は事前に真のPを知ることができないので,サンプルサイズの変化に伴う実行時間の傾向のみを観察することが最大の関心事である.
特に括弧内は、すべての生物学的複製でゼロを持ち、一方のグループでゼロ平均、他方でゼロ平均より大きい特徴、すなわち構造的ゼロを考慮する各方法が採用する根拠に値する(表1の4列目を参照)。DA特徴を用いた主なシミュレーションフレームワークでは、これらの分類群に対して各手法が操作した選択肢を含む手法の精度と想起を計算しました。これらのケースをどのように考慮するかによって手法の評価に偏りが生じないようにするため、これらの分類群をTNとして扱う追加調査を行った(S1ファイルの図6-17参照)。
結果
DAタクサを含まないシミュレーションにおける誤検出率の抑制
図3は、2つのデータセットに含まれるすべての分類子をDAではないとシミュレーションした場合の、SSとデータセットが異なる場合の各シミュレーションの平均FPRです。
拡大する
図3. DA featureをシミュレートしないシナリオでの比較で考慮した各データセットに対する各差分アバンダンス法の偽陽性率(FPR)。
データセットに対応する各ボックスでは、ツールは行に、異なるサンプルサイズ(SS)値は列に表示されている。FPR 値は 50 回のシミュレーションの平均値であり,棒グラフは標準誤差を示す.ANCOMラベルは、基礎となるFDR調整を行わずに実行した方法を意味します。
doi:10.1371/journal.pcbi.1010467.g003
詳細 "
ANCOMは,DAラベルの割り当てに使用するw統計量分布を定義するためにFDR制御手順を利用する(S1ファイルのセクション2).FPRは他の手法の非調整p値で計算されるため、基礎となるFDR調整を行わない手法も実行した(ANCOM)。
タイプIエラー率は、すべての方法について、考慮された各SSに対して実行されたFPRの良好な制御を実証しています。edgeRだけがすべてのデータセットで常に閾値をわずかに超える傾向があり(平均FPR値は0.1未満ですが)、他のツールは平均して望ましい限界に達しています。SS = 10の歯のデータセットでは、ANCOM-BCも低いパフォーマンスを示しています。実装された2つのANCOMのバリエーションに顕著な違いがないことは興味深いことです。したがって、w統計量を得るための対数比検定の補正は、タイプIエラーの抑制に影響しないようです。
予想通り,サンプルサイズの共変量は,FPRの点では各手法の性能に大きな影響を与えない.しかし、ANCOMとANCOM**は、SS = 25からFPRの過剰制御を達成した。ALDEx2もFPRが5%の閾値より低くなっていますが、この挙動はすべてのサンプルサイズ構成で一定しています。したがって、ANCOMとALDEx2は、タイプIエラーを制御する上で最も保守的な方法であることがわかる。
上記の考察は、異なる実行における FPR 値の分布を観察しても有効である(S1 ファイルの図 18 を参照)。
DAタクサを用いたシミュレーションの性能評価
この段落では、θ=1-f2で低存在のDA特徴の導入を回避する、主なシミュレーションフレームワークの結果について説明する。
誤発見率、誤検出率。
FDR値が低いということは、DAと同定された特徴の中でFPが少ないということである。つまり、その手法は、発見された特徴の中からDA特徴を正しく選択していることになる。ほとんどの手法はFDRを制御する手順を用いているため、この指標の期待値は設定された閾値付近、つまり我々の評価フレームワークでは0.05である。
図4は、比較対象とした各データセットにおいて、主な共変量であるサンプルサイズとDA特徴の割合の組み合わせごとに、50回のシミュレーションで平均したFDR値を示しています。
拡大する
図4. メインシナリオの比較で考慮した各データセットについて、シミュレートしたDA特徴を用いた各ディファレンシャルアバンダンス法の偽発見率(FDR)。
データセットに対応するボックスの各セットでは、シミュレートされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)値が列に表示されている。FDR値は50回のシミュレーションの平均値で、棒グラフは標準誤差を示す。FDRの定義値を提供する実行回数は、棒グラフの最初に示されている。
doi:10.1371/journal.pcbi.1010467.g004
詳細 "
しかし、サンプルサイズが小さい場合、ANCOM、ANCOM-BC、corncob_wald、edgeR、DESeq2では、その効果は望ましい閾値を下回るほどではなく、metagenomeSeqとcorncob_lrtではP=5のみである。3つのデータセットにおける各手法は同等の平均値を示しており、シーケンスの深さは主に影響を与える共変量ではないことが示唆されました。
詳細には、SS = 50とSS = 100の場合、各手法は期待値に達するか、時にはわずかに上回る傾向があります。しかし、Corncob、DESeq2、edgeRは、多くのシナリオでFDRをコントロールできない傾向があります。一方、サンプルサイズが小さい場合、ALDEx2、eBay、Wilcoxon、MaAsLin2は、通常他のツールは確実に失敗する傾向がある中で、良いパフォーマンスを維持しています。しかし、SS = 10の構成は、多くのシミュレーションが不定精度を持つため、あまり有益ではありません。つまり、この方法は、どの特徴量も差分として識別しないのです。
実際、SS = 10でのFDR値の分布(S1ファイルの図19参照)を見ると、metagenomeSeq、ANCOM、ANCOM-BC、corncob_lrtのような固有のばらつきがある手法と、edgeRやcorncob_waldのように精度値の数が利用できないことによるばらつきを特徴とするものが存在することがわかります。
構造的なゼロをDAとみなすANCOMとANCOM-BCは、SS=10と25のときに明らかに性能が低下している。明らかに、構造的なゼロをTNとして評価する場合、つまり、一方のグループの平均がゼロで、他方のグループの平均がゼロより大きい特徴を考慮する場合、ANCOMとANCOM-BCはSS = 10とSS = 25で性能が向上する(S1ファイルの図20と21)。しかし、このシナリオでは、他のすべての方法は、目に見える形で性能を変更することはありません。
一方、FPRはすべてのシミュレーションにおいて、すべての手法で5%以下に抑えられている(S1ファイルのFig.22-25)。したがって、前項で得られた傾向が確認されたことになる(Fig 3)。
リコール
高い再現性は、シミュレーションされたDA分類群のほとんどが手法によって正しく分類されたことを意味する。したがって、高い再現性を実現する手法が望まれる。共変量のすべての組み合わせについて、手法の性能をS1ファイルの図26と図27にまとめ、リコールとFDRの両方を図5に示す。
拡大する
図5. メインシナリオの比較で考慮した各データセットについて、模擬DA特徴を用いた各差分豊度法の平均Recall(y軸)およびFDR(x軸)。
データセットに対応するボックスの各セットでは、シミュレートされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)値が列に表示されている。リコール値は50回のシミュレーションの平均値で、棒グラフは標準誤差を示す。
doi:10.1371/journal.pcbi.1010467.g005
詳細 "
一般に、すべての方法は、SSが増加するにつれて、リコールがかなり改善することを示す。共変量が異なると、手法のランキングに影響があるようです。歯のデータセットでは、SSが50から100に増加すると、corncob_lrt、corncob_wald、DESeq2、edgeRが顕著にリコールを増加させる。一方、他の方法は、一般的にWilcoxonベースのアプローチに匹敵する低いリコールで特徴付けられます。一方、同じ構成のSSを見ると、BFとnonIBDでは、ほとんどのシナリオでeBay、ANCOM、ANCOM-BC、DESeq2、MaAsLin2が最高のパフォーマンスを達成している。しかし、手法間の順位はそれほど顕著ではなく、想起値はウィルコクソンのベースライン手法で得られた結果から大きく逸脱することはありません。
サンプルサイズが小さい場合は、ANCOMとANCOM-BCが考慮すべき方法であると思われるが、SS = 10とSS = 25のリコール値が特に低く、不満足であると考えられることは明らかである。
実験グループあたりのレプリカ数を多くすることで再現性が向上することが知られているが、この結果から、望ましい数は50以上であり、μの左寄りの分布が大きいデータセット(BFやnonIBDなど)においてのみ100以上と考えられる。
MetagenomeSeqは、検討するデータセットにかかわらず、ほとんどのシナリオで他の方法よりも低いリコール値という特徴があります。
分析者は通常、FDRの有意閾値を0.05に設定し、FDRの期待値をコントロールしながらより高いRecallを達成する方法を知ることに関心があります。図5に示すように、SS = 100の歯のデータセットでは、DESeq2、corncob、edgeRは、ほとんどのシナリオでFDRを望ましい有意水準(すなわち黒線)で制御しながら、最高のRecall値を達成するので、最良の方法であることがわかります。しかし、他のデータセットを見ると、明確に定義された勝者はいない。一般に、サンプルサイズが小さい場合、いくつかの方法はまだFDRを制御していますが、想起の観点からの性能は極めて低いです。
最後に、ヌル分類群の特殊なケースをTNとして扱っても、リコール指標に影響はありません(S1ファイルの図28と29)。
Precision-Recallのトレードオフ。
図6は、50回のシミュレーションの値を平均して、すべての構成で手法によって得られたAUPRを示す。サンプルサイズが大きくなるにつれて、特にSS = 10からSS = 25の間で面積の値が大きくなっている。むしろ、DA特徴の割合は、サンプルサイズが小さいほどAUPRに顕著に影響する。
拡大する
図6. メインシナリオの比較で考慮した各データセットについて、模擬DA特徴を用いた各差分豊度法のASPR(Area Under Precision-Recall Curve)を示す。
データセットに対応するボックスの各セットでは、シミュレートされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)値が列に表示されている。AUPR値は50回のシミュレーションの平均値で、棒グラフは標準誤差を示す。
doi:10.1371/journal.pcbi.1010467.g006
詳細 "
一般に、明確に定義された順位はなく、各手法はかなり比較可能である。主に、SS = 50またはSS = 100の歯のデータセットでは、corncob_lrt、corncob_wald、DESeq2、edgeRのAUPRはわずかに高い。一方、ANCOMは、低いサンプルサイズに対して低いパフォーマンスを示し、いくつかのシナリオでは、Wilcoxonベースのアプローチよりもさらに低くなります。しかし、SSの増加により、他の手法と同程度の結果まで戻っています。さらに、ALDEx2は、BFとnonIBDデータセットでは概ねランキング1位の値を示しているが、Toothではそうではない。しかし、ALDEx2は低いサンプルサイズでもAUPRの平均値をかなり維持しており、edgeRのように低いサンプルサイズでもランキングの上位を維持できない手法があるのとは対照的である。
また、異なる実行における変動に関しても、テストされた異なる構成間でメソッドは非常に同等です(S1ファイルの図30を参照)。
S1ファイルの図31と図32に、1つのシミュレーションにおける平均PRカーブとPRカーブの例を示しています。ANCOMとANCOM-BCは、SS = 10とSS = 25において、低い値から始まる曲線を示し、他の方法と異なる傾向を示した。
最後に、pAUPRは、以前発見されたことを補強しています(S1ファイルの図33と34)。高いサンプルサイズ(基本的にSS = 50とSS = 100)では、AUPRのほとんどが精度値の1~0.9の間の区間に収まっています。その代わり、すでに低い性能を示している他の構成では、望ましい範囲に含まれる部分が極めて小さくなっている。
平均AUPRのパフォーマンスは、構造的なゼロをTNとみなす調査(S1ファイルの図35と36)、特にSS = 10とSS = 25で上昇するようで、corncobとDESeq2は別で、閾値を変更しても特徴のランキングがより安定していることがわかる。上記の観察は、pAUPR(S1ファイルの図37と38)についても有効であり、構造的なゼロのためにANCOMとANCOM-BCでのみ一貫して増加します。実際、PR曲線で観察された上記の傾向はかなり減少し、ANCOMのSS = 25でのみ確認できます(S1ファイルの図39と図40)。
データセット特性の違いによる手法の性能への影響
モデルやシミュレーションフレームワークのパラメータ、およびデータ特性の変化を考慮し、手法の性能に関する概観を完成させた。特に、同一データセットにおける元の配列深度ベクトルの減少や増加、条件間の配列深度のアンバランスを調査する。そして、特徴のばらつきが減少したシナリオで得られた結果について議論する。さらに、存在量の少ない複数のDA特徴が存在する場合に、信頼性の高い解析を行う手法の能力について扱う。最後に、正規化の効果を検証し、異なる生態系ニッチに由来するデータセットで調査を拡張する。
シーケンスの深さによる影響
非IBDデータセットでシーケンシング深度パラメータLを変化させて得られたシミュレーションに関するすべてのメトリクスの結果を、S1ファイルの図41〜52に示す。このタイプの調査は、同じ生物学的現実を調査した場合、シーケンシング深度の大小が手法の性能に影響を与えるかどうかを理解することを目的としています。
その結果、μとφが同じであれば、Lの変化だけでは手法の出力に大きな影響を与えないことが確認されました。この現象は、RNAシーケンスの分野ですでに観察されている[42,43]。
アンバランスな配列深度シナリオでは、やはりバランスシナリオと同様の傾向が観察される(S1ファイルの図53~64を参照)。しかし、FDRは、高サンプルサイズ、すなわちSS = 50とSS = 100におけるDAの割合の変化により影響を受けるようである。特に、ALDEx2、ANCOM、eBay、ANCOM-BCは、P = 20とSS = 50-100でFDRを制御できず、eBayはSS = 25でもすべてのPでFDRの増加を示した。一方、metagenomeSeqとMaAsLin2は、FDRを最も制御できるツールで、これもP割合に影響を受けない(ただしリコールは最も低いまま)。AUPR値を見ると、バランスの取れたシナリオに関して変化は見られません。しかし、ALDEx2、eBay、ANCOMでは、SS = 100とP = 5の場合のみpAUPR値が低くなっています。他のシナリオでは、手法のランキングは維持され、pAUPRの値も同等になっています。
特徴のばらつきの影響。
ばらつきの低減は、真の個体群がガンマ分布でシミュレートされた平均絶対存在量付近での分散が少なくなることを意味します。その結果、分散の減少により、2つの実験グループにおける単一分類群の分布の差が大きくなる可能性があります。その結果、配列決定プロセスによってもたらされるノイズが同じであれば、DA特徴を正しく認識する確率が高くなる可能性があります。しかし、理論的には想起率の向上が期待されるとしても、その重みは不明である。
S1 File の Main Fig 7 と Fig 65 は、"recall "を設定したときの結果である。BFデータセットと非IBDデータセットでは、望ましいSSの数はそれぞれ50以上、100以上である。再び、歯のデータセットでは、想起値はより高く、SS = 25からcorncob_wald、DESeq2、edgeRが優れている傾向があり、明確な傾向が見られる。SS = 100で他の2つのデータセットを観察しても、同じ結論が導き出される。しかし、この場合、サンプルサイズが小さい場合(SS = 25とSS = 50)、eBay、ANCOM、ANCOM-BC、MaAsLin2、edgeRの中で勝者を特定することは困難です。
拡大する
図7. 変動性を抑えたシミュレーションにおける、比較対象とした各データセットに対する各Differential Abundance法の再現性。
データセットに対応する各ボックスのセットでは、シミュレーションされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)値が列に表示されている。想起値は50回のシミュレーションの平均値で、棒グラフは標準誤差を示す。
doi:10.1371/journal.pcbi.1010467.g007
詳細 "
各手法で同定されたTPの数は増加しているが、FDRの制御(S1ファイルのメイン図8と図66参照)は、図4で見られたものと同様である。特に、多くのシナリオでFDRをうまく制御しているmetagenomeSeq、eBay、ANCOMを除き、いくつかの手法ではSSが増加すると閾値に到達したり超過したりする傾向があることがわかります。
拡大する
図8. 変動性を抑えたシミュレーションにおける、比較対象とした各データセットに対する各Differential Abundance手法のFDR。
データセットに対応する各ボックスのセットでは、シミュレーションされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)の値が列に表示されている。FDR値は50回のシミュレーションの平均値で、棒グラフは標準誤差を示す。変動幅を小さくしたシナリオと、シミュレートされたDAの特徴を持つメインシナリオの間の平均FDRの差は、棒グラフの最初に示されています。
doi:10.1371/journal.pcbi.1010467.g008
詳細 "
この場合も、FPR値は予想される閾値を下回っている(S1ファイルのFig.67と68)。最後に、AUPRとpAUPRのメトリクスに関する結果は、一般的に、方法間で観察された差は減少する傾向にあるものの、以前に行われた評価に従っている(S1ファイルの図69~74)。
低存在率の分類群の影響
本シミュレーションでは、閾値の除去、すなわちθ=0は、低存在量であってもDA特徴の挿入を意味する。前述したように、この範囲ではノイズの存在が大きくなるため、性能の低下が予想される。
実際、BFとnonIBDのデータセットでは、SS = 100でもリコール値が特に低い(S1ファイルのメイン図9と図75を参照)。一方、歯のデータセットでは、サンプルサイズが大きくなるにつれて、corncob、DESeq2、edgeRが他の手法を上回る傾向が明確になっていることがわかる。BFデータセットとnonIBDデータセットで性能が低いのは、左寄りのμ分布が多いためと考えられます。その結果、DAとしてシミュレートされる極端に少ない量の特徴の数が、歯のデータセットよりも多くなる可能性がある。したがって、BFとnonIBDは、シーケンシングノイズによって不明瞭になったDAフィーチャーの数が多いという特徴を持つ可能性があります。
拡大する
図9. 図9: シミュレートされたDA特徴量とθ = 0のシナリオにおける、比較対象とした各データセットに対する各差分存在量法のリコール率.
データセットに対応する各ボックスでは、シミュレートされたDA特徴の異なる割合(P)が行に、異なるサンプルサイズ(SS)値が列に表示されている。想起値は50回のシミュレーションの平均値であり、棒グラフは標準誤差を示す。
doi:10.1371/journal.pcbi.1010467.g009
詳細 "
各手法は、特にP = 15とP = 20の場合に、歯のFDRをうまくコントロールしています(S1ファイルの図76と77)。しかし、他のデータセットでは、ほとんどのシナリオでmetagenomeSeq、MaAsLin2、ALDEx2を除いて、期待限界値を超える傾向がわずかに強調されている。
これらの結果を総合すると、本手法はFPを良好に制御して低存在DA特徴を検出することができるが(S1ファイルの図76~79)、リコール(S1ファイルのメイン図9と図75)および全体の精度-リコールトレードオフ(S1ファイルの図80~85)が低下していることがわかる。
正規化の効果。
S1 FileのFig.86-91は、DA分類群が中高域にあるシナリオにおいて、GMPR正規化を用いて得られた結果である(すなわち、θの閾値を設定することにより)。図10と図11は、検討中の3つのデータセットのうちの1つについてのFDRとRecallの例を示し、各手法の正規化バージョンと非正規化バージョンの間の統計的有意差(ウィルコクソン検定、多重検定で補正しない公称p値が0.05より低い)は、左側の星印で示されている(他のデータセットについてはS1ファイルの図92と93を参照)。FDRとRecallの差は、手法やデータセットによって、符号も大きさも異なることがわかる。しかし、ほとんどの差は統計的に有意ではなく、基本シナリオと比較して、GMPR正規化によって明らかに利益を得るツールはありませんでした。全体として、GMPR正規化は、基本シナリオ(デフォルトの正規化で実行されたメソッド)と比較して、FDRとRecallの両方の観点から、メソッドのランキングに明確な影響を及ぼさない。
拡大する
図10. DA featureをシミュレートしたシナリオの歯のデータセットについて、各差分存在量法とそのGMPR正規化版の間の平均FDR差[%]。
行にはシミュレートされたDA特徴の割合(P)、列にはサンプルサイズ(SS)の値を示している。各行の先頭の数字は、デフォルトの正規化で得られたFDR値に対応し、記号()はウィルコクソン非対称統計検定が有意であることを示す。
doi:10.1371/journal.pcbi.1010467.g010
詳細 "
拡大する
図11. 図11 DA特徴をシミュレートしたシナリオの歯のデータセットにおける、各ディファレンシャルアバンダンス法とそのGMPR正規化版の平均Recall差[%]。
行にはシミュレートされたDA特徴の割合(P)、列にはサンプルサイズ(SS)の値を示しています。各行の先頭の数字は、デフォルトの正規化で得られたRecall値に対応し、記号()は、ウィルコクソンのペア統計検定が有意であることを識別するものである。
doi:10.1371/journal.pcbi.1010467.g011
詳細 "
非ヒト関連ニッチによる影響
異なる生態系ニッチを考慮すると、一般的な傾向は有効であるものの、FPR、FDR、想起において異なる絶対性能が観察された(S1ファイルの図94~105を参照)。一般的に、いくつかの例外を除き、ツールはFPRを0.05以下に維持できることが確認された。例えば,2つのcorncob variantは,SS = 100とP = 15, 20で0.05よりわずかに高いFPRを示した.同様に、edgeRとWilcoxはSoilデータセットでP = 20のときにFPの数が多くなっています。一方,MetagenomeSeqはFPRの点では良好な結果を示しているが,他の手法と比較してRecallは最も低い.
ヒトに関連するデータセットでは、FDRはFPRよりも制御が難しい手法もある。これは、非人間関連環境の場合、SS = 50とSS = 100で特に顕著で、FDRが最大25%に達する手法もあります。しかし、FDRを正しく制御できる手法は、ヒトと非ヒトのデータセットで同じである。
ヒト関連データセットとは異なり、AnimalGutデータセットのmetagenomeSeqを除き、ほとんどのメソッドが低いサンプルサイズでも少なくとも1つのDAフィーチャーを呼び出すことが可能である。一貫して、リコール値はヒト関連データセットに対して増加し、大半のメソッドはSS = 25で既に満足のいく値(約75%)を示しています。
総合的な性能のまとめ
生態系ニッチは16S rDNA-seq研究において容易に入手できる情報であり、我々の研究でもDA法の性能に影響を与える特徴であるが、特徴の変動、DA分類群の割合、低存在DA分類群の量は、研究者が通常コントロールできない、あるいは事前情報がほとんどない要素である。
一方、サンプル数、シーケンス深度、カウントの正規化は、16S rDNA-seq研究中にコントロールできる要素であるが、サンプル数のみがDA解析にかなりの影響を与えるように思われる。
そこで、DA解析の精度に最も影響を与えると思われる要素、すなわち生態学的ニッチとサンプル数に基づいて、手法の結果をまとめ、特定のシナリオにどのDA手法がより適合するかについての推奨事項を提示することにしました。我々は、シミュレートされたDAが中高の範囲にあるシナリオ、すなわちθの閾値を設定した場合の5つのデータセットにおけるメトリクスの値を比較することにした。基本的に、主に低存在域のDAをシミュレートしてこの比較を行うことは、メソッドのパフォーマンスが劇的に低下することが既に確認されているため、意味がない。
FDR、Recall、少なくとも1つの分類子がDAと同定されたデータセットの割合(NA_perc)、およびDA分類子の割合(P)を組み合わせて全シミュレーションで計算したpAUPRの平均値は、未知の共変数でありほとんどのシナリオで特に指標に影響しないため考慮しない。計算時間は,我々の設定ではシングルコアでツールを実行しても無視できる程度であるため,考慮していない(ただし,並列計算をサポートしているものもある。)
S1ファイルの図106と図107は、すべてのメトリクスの平均値を示しており、各メソッドの平均正規化ランキングをカラースケールで報告しています。データセットが変わると絶対的な平均値は変化するが、データセットには2つの異なるグループが存在する。BF、nonIBD、Soilは類似のパターンを持ち、すなわちALDEx2、ANCOM-BC、DESeq2、eBayはすべてのメトリクスを考慮してトップダウンランキングに入っている。一方、toothとAnimalGutは、corncob_wald、corncob_lrt、DESeq2、edgeRで同様のトップダウンランキングのパターンを示している。まとめると、生態学的ニッチは、絶対的な性能に影響を与える一方で、人間と人間以外の関連データセット間で同様のパターンが観察されるため、手法の全体的なランキングに影響を与えないようです。
しかし、S1ファイルの図106と図107に示したランキングは、メトリックの平均値を考慮していない。例えば、すべてのメソッドがFDRを制御できなかった場合でも、ランキングはメソッドの中から勝者を見つける。そのため、メトリクスの値にいくつかの閾値を定義することによって、メトリクスの各平均値に1~5(1=「低」、2=「低中」、3=「中」、4=「高中」、5=「高」)のスコアを割り当てます。簡単に言うと,精度が高く,公称FDRを0.05に抑えた手法(すなわち,精度>=0.95)を "high "クラスに割り当てた.0.90を超える精度を持つメソッドは、"high-mid "クラスに分類され、非常に高い精度を持ちながら、選択したFDRの閾値をわずかに超えるツールを表しています。全体として、上記の2つのクラスのメソッドは、タイプIエラーをコントロールすることが研究に関連する制約である場合に、ロバストな選択肢を表しています。残りのクラスは、0.9未満の精度を持つメソッドで、「低」クラスには最大精度0.5を達成したメソッドが含まれています。精度と密接に関連して、我々は「NA_perc」、すなわち少なくとも1つの分類子がDAとして同定されたデータセットの割合の点でも方法を分類した。大多数のデータセットで少なくとも1つのDA分類子を同定できる手法(75%以上)は、最も高いクラスに分類された。特に、100%もしくは90%以上、75%以上のデータセットで少なくとも1つのDA分類子を同定できた手法は、それぞれ「high」、「high-mid」、「mid」のクラスに割り当てられた。リコールに関する性能は、リコール範囲[0-1]の5分位を表す5つのクラスでランク付けされた。回収率は、異なるサンプル数SSで最大の差を示す指標であり、ほぼ全範囲[0-1]をカバーするため、5つの分位で層別化することで、メソッドのパフォーマンスを要約する公正でわかりやすい方法を提供します。pAUPRに関しては、最も低いクラスはpAUPR < 0.5を持つメソッドを表し、pAUPR > 0.7は0.1ステップで「高」、「高-中」、「中」とラベル付けするのが妥当である。
図12は、各データセットのすべてのSSシナリオにおいて、上記のようにランク付けされた手法を、最初に精度、次に再現性でソートして示している。SS=10では、MaAsLin2が常に精度をコントロールし、5つのデータセットのうち2つでより高いリコール値に達しています。サンプルサイズが大きくなるにつれて、あるパターンが明確になるようです。ALDEx2は、SS = 25、50、100のすべてのデータセットでFDR制御の点で最良であり、同様にeBayはSS = 25-50で良い結果を示し、ANCOMはSS = 50-100で最高の性能を達成した。これらの手法のリコール値は上位に位置しているが、大半のデータセットでFDRを制御しないことを犠牲にして高いリコールを達成する手法もある。
拡大する
図12. 各DA手法の総合性能。
異なるサンプルサイズ(SS)値に対応する各セットのボックスにおいて、列には各データセットのPrecision、NA_perc(利用可能な精度の割合)、RecallおよびpAUPRスコアが示されている。メソッド(行)は、すべてのSSシナリオのPrecision値に基づいてランク付けされ、同点の場合はRecall値に基づいてランク付けされます。ボックスの下の凡例では、各メトリクスの総合スコアを割り当てるために使用した閾値を説明しています。
doi:10.1371/journal.pcbi.1010467.g012
詳細 "
ディスカッション
本研究では、サンプルサイズとDAの割合が与えられた2つの実験条件でマイクロバイオームデータセットを生成することができる、信頼性の高いシミュレーションフレームワークを利用しました。また、既知のDA分類群の存在量表を作成することで、この研究分野で使用されている主要な手法を幅広く評価することができる。特に、metaSPARSim [23]によって導入されたγ-MHGモデルに基づくシミュレーションフレームワークを利用しました。これは、16Sデータの組成的性質を再構成する優れた能力を示し、性能に対するパラメータの影響を調べることができ、評価に関与するツールがこのモデルを想定していない(偏りの可能性を回避)ためです。最近、他のシミュレーションツールが文献で提案された: MB-GAN [44]とSPARDOSSA2 [45]である.前者は、深層学習研究[46]の生成的敵対ネットワーク(GAN)に基づくフレームワークを利用しており、DA機能をシミュレーションするためのパラメータ設定方法は明確になっていない。一方、SPARDOSSA2は、分類群間および分類群と環境との相互作用の構造を考慮した階層型モデルを提案しています。後者の特徴は一般的に注目されているものですが、シミュレーションの段階で分類群間の相関構造を考慮しても、一変量DAアプローチには直接影響しないはずです。さらに、SPARDOSSA2はショットガンデータでのみテストされており、16Sカウントデータといくつかの特徴を共有しているものの、データ分布の違いにつながる可能性がある分類学的解像度の点でいくつかの違いがある[47-49]。例えば、Calgaroら[19]は、16Sデータセットに最も適合する分布は負の二項分布であるが、ショットガンデータはゼロ膨張負の二項分布によって最も適合すると結論付けている。文献では、Kurtzら[51]が提案したNormal-to-Anything[50](NorTA)ベースのアプローチや、Prostら[52]のゼロ膨張多変量ガウスコピュラモデルなど、カウントテーブルをシミュレーションするための他のアプローチもある。しかし、これらのアプローチはネットワーク再構築法の分野で提案されたものであり、16sデータ分布の再現能力については広く評価されていない。
性能評価は、データセットの生物学的および技術的特性の両方に関連するシミュレーションパラメータの影響を考慮して実施された。特に、グループ間のDA特徴の定義は、強度に応じて異なるFCの範囲を適用する非一定FCアプローチに基づき、生物学的データで観察される強度-変動性の関係を保証しています。さらに、DA特徴の割合(P)、実験グループの被験者数(SS)、シーケンスの深さ、正規化方法、生態系ニッチなど、すべての共変数の組み合わせをテストする広範な評価を実施しました。
テストした手法の中には、ほとんどの特徴がDAでないと仮定しているものがあります。そのため、Pの数を特に多く設定すると非現実的となり、手法の性能に大きな影響を与える可能性がある。また、我々のフレームワークでは、DA分類群の数は、平均μがθより大きい特徴の数に依存する。BFデータセットでは、この制約を満たす特徴の割合が20.5%であるため、BFではθ=1-f2のとき、高い値ほど可能なすべてのDA分類群を挿入することになることを考慮して、P = 5%-15%-20% とした。SSの値については、文献[11,15,17-19]で検証された傾向に従い、[10,25,50,100]のSSを選ぶことにした(S1ファイルの表Bを参照)。この値は、サンプル数が限られているパイロット試験から、コホートがより大きい完全な実験または観察研究まで、異なる研究シナリオを表していることに留意されたい。
我々の結果は、タイプIエラーをうまくコントロールすることを確認した。SS=50とSS=100の場合、一般的にすべての手法が期待値に達するか、時にはわずかに上回る傾向があることが確認されました。ALDEx2、eBay、および2つのベースライン手法は、低いサンプルサイズでもFDRの面で良好な性能を維持しています。corncob_lrt、corncob_wald、DESeq2、edgeRはtoothで高い再現性を示し、eBay、ANCOM、ANCOM-BC、DESeq2はBFと非IBDデータセットで高い再現性を示しています.Wilcoxonの性能は、特に中心対数比変換を行わない場合、他の手法と同等であり、ノンパラメトリック検定が価値あるアプローチである可能性を示唆しています。
Hawinkelら[17]でも、edgeRとDESeq2に関するいくつかの例外を除き、タイプIエラーに関する同様の知見が得られています(いくつかの構成で)。また、Calgaroら[19]では、我々の比較に含まれる多くのメソッドは、ALDEx2を除いて、平均FPR値が閾値をわずかに超えており、正しく制御されています。最後に、Khomichら[20]は、彼らの比較に含まれるすべてのメソッドで0に近いFPRを観察しました。
Hawinkelら[17]と同様に、我々の結果は、サンプルサイズを大きくすることで、想起の面で性能が向上することを示していますが、FDRの制御については明確な傾向が見られません。Linら[13]は、DA特徴の割合を増やすと、想起値に大きな影響を与えずに、FDRの制御を改善する傾向があることを確認しています。
一般的に、Differential Abundanceテストにおける正規化の効果については、文献上では意見が分かれている。Hawinkelら[17]は、手法のパフォーマンスを大幅に向上させる正規化は存在しないと報告しています。したがって、ツールの著者が提案するデフォルトの正規化、またはシーケンス深度補正の単純な使用はどちらも妥当な選択です。最近、Baruzzoら[53]は、正規化が差分テストに良い影響を与えることを観察したが、その効果量は限られている。Linら[13]で示されたパフォーマンスも、異なる正規化方法が想起に限定的な影響を与えるが、精度にはより明白であることを示している。実際、Linら[13]では、3つのシミュレーションシナリオの1つでFDR制御の結果を変更するように見えるテストされたものの中で唯一の方法は、正規化なしのバリアントは、TSS正規化で得られたものと比較して、高い回収と低いFDR値を有するウィルコクソンテストです。Mallickら[29]では、TSSは、モデルベースの正規化スキーム(RLEなど)またはデータ駆動型の正規化手法(TMM、CSS、ANCOM、ALDEx2、eBayの対数比変換など)と比較して、FDRコントロールへの影響と優れた統計力の観点から、最適な正規化の選択肢として特定されています。最後に、Calgaroら[19]では、正規化は、特に一貫性(すなわち、独立したデータで再現可能な結果を生成する各手法の能力)および差分存在量分析のフレームワークの観点から推論結果に影響を与えるようです。ベンチマークで得られた結果は、Hawinkelら[17]の結果と同様に、GMPRが総合ランキングに影響を与えないことを示しています。
過去のベンチマーク研究における総合的なパフォーマンスを見ると、類似点と非類似点が見られますが、これはおそらく使用するシミュレーションフレームワークが異なるだけでなく、調査対象の手法の実装やパラメターが異なることに起因していると考えられます。例えば、Hawinkelら[17]では、metagenomeSeqとedgeR以外の手法は、高いサンプルサイズでも極めて低いリコール値を示しています。ALDEx2だけがFDRを制御しているように見えますが、DESeq2、edgeR、metagenomeSeq、ANCOMはほとんどのシミュレーションシナリオで公称値を超えています。一方、Linら[13]は、ALDEx2がP = 15とP = 25でそれぞれ5%の閾値を下回るかわずかに上回るものの、ANCOMとANCOM-BCだけがFDRの点で合理的に機能すると結論付けています。DESeq2、edgeR、Wilcoxon(TSS正規化あり)は、ANCOMやANCOM-BCと同等の高い想起値を維持しながら、FDRの観点からは低いパフォーマンスを示しています[13]。さらに、Linら[13]は、ALDEx2が競合するDA手法と比較してリコール値が小さいことを発見したが、一般的に、その値は、同様のサンプルサイズ値について以前のベンチマーク研究で観察された値よりも高いものである。最後に、Khomich ら [20]は、DA 分類群の割合が低い場合、ANCOM とは対照的に DESeq2 と ALDEx2 は想起値の面で劣ると結論付けています。
研究者は、DA 分類群の誤認を防ぐことに第一に関心があるため、私たちの研究では、FDR の制御を全体的な性能評価の主な指標と考え、結果を要約するようにしています。その結果、低いサンプルサイズSS = 10の場合、MaAsLin2は、低いリコールではあるが、常にFDRを制御していることがわかった。一方、他のシナリオでは、ALDEx2、eBay、ANCOMが最適なアプローチであると思われます。同様に、Nearingら[54]では、ALDEx2とANCOMがデータセット間で一貫した結果を示しただけでなく、異なるアプローチと最もよく一致した。さらに、データセットにおける性能のばらつきを考慮し、著者らはコンセンサスアプローチを推奨しています。我々の発見は、焦点を当てるべき最適なツールを特定するのに役立つ。
metagenomeSeqに関する我々の結果は、過去の比較と一致しないことは注目に値する。特に、他の研究[13,17,19]では、metagenomeSeqは高いリコールと高いFDR値によって特徴付けられるが、我々は逆の挙動を観察した。実際、metagenomeSeqは、zero-inflated Log-Gaussian(ZILG)混合モデルとzero-inflated Gaussian(ZIG)モデルという2種類のモデルを実装しています。ZILGモデルはZIGモデルと比較して非常に高い性能向上を示しているため、著者らの提案に従うことをお勧めします(2つのアプローチの比較はS1ファイルのセクション9に示されています)。
我々のアプローチの限界は、比較した方法がデフォルト設定で実行されていることである。デフォルト設定に対して異なる実装を選択した場合の性能への影響をより理解するためには、さらなる調査が必要である。一方、実際のアプリケーションでは、デフォルトのパラメータを持つツールを使用することが最も一般的な選択です。もう一つの限界は、平均的な真の絶対量または相対量における有意差の特定を目的とした一変量法に焦点を当てたため、差分ランキングアプローチ [14] や、観察された差をもたらす分類群の亜集団またはグループの特定を目的とした gneiss [55] または selbal [56] を除外したことです。もう一つの疑問点は、他の研究で行われたように、我々は同じ数の被験者のグループを持つデータセットで手法の性能を比較したことです。2つの実験グループで異なるサンプルサイズを設定してさらに比較することは、文献でよく見られるシナリオであるため、確かに有用であろう。最後に、異なる微生物相ニッチ間の比較で指摘されたように、異なるDA手法の相対的なランキングは非常に安定しているように見えるが、結果はデータセットのタイプや、異なるタイプのサンプル調製、ライブラリ、シーケンスプラットフォーム、読み取り前処理から生じる特徴に依存するかもしれない。実験的な共変量に対応したより広範な比較は本稿の範囲外であるが、今後の研究の興味深い展開として、DA法がその性能と分析対象データセットの実験的特徴に基づいてどのようにクラスタリングされるかを分析することに焦点を当てることができる。
シミュレーションのシナリオによって性能がどのように変化するかを観察することで、重要な結論を導き出すことができます。特に小さなサンプルサイズ、例えばSS = 10では、多くのメソッドがDA特徴を見つけられない傾向にあり、その結果、リコールはゼロ、FDRは未定義となる。これは研究設計の段階で考慮すべき点である。平均絶対量分布の違いは、おそらく想起性能にマイナスの影響を与える。また、特徴量のばらつきが少ないと、想起性能にプラスの影響を与えることが分かっています。したがって、強度-可変性の関係も手法の性能に影響を与える可能性がある。この側面は、ベンチマーク研究だけでなく、DA解析のための新しいツールの開発においても考慮されるべきものである。
本研究の結果は、存在量差分解析のためのバイオインフォマティックツールの改良にさらなる努力が必要であることを示すものである。本研究は、新しい技術や解析方法の継続的な開発を考慮すると、将来の比較研究のための基盤を強化したと考えている。実際、私たちが提案したシミュレーションの枠組みは、真の絶対量の平均と分散を含む他のパラメトリックモデルに拡張することが可能である。さらに、我々の研究は、16S rDNA-Seqカウントデータのための特定のツールの信頼性と品質をさらに向上させるために、共通の評価フレームワークを開発し使用する科学コミュニティを支援することを目的としています。
参考情報
参考文献
1.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019 Aug;178(4):795-806.e12. doi: 10.1016/j.cell.2019.07.008. pmid:31398337.
2.Daisley BA, Chanyi RM, Abdur-Rashid K, Al KF, Gibbons S, Chmiel JA, et al. Abiraterone acetateは、去勢抵抗性前立腺がん患者において腸内常在菌Akkermansia muciniphilaを優先的にエンリッチします。Nat Commun. 2020 Sep;11(1):4822. doi: 10.1038/s41467-020-18649-5. pmid:32973149.
3.Berbers R-M, Mohamed Hoesein FAA, Ellerbroek PM, van Montfrans JM, Dalm VASH, van Hagen PM, et al. Low IgA Associated With Oropharyngeal Microbiota Changes and Lung Disease in Primary Antibody Deficiency. Front Immunol. 2020 Jun;11:1245. doi: 10.3389/fimmu.2020.01245. pmid:32636843.
4.Edslev SM, Olesen CM, Nørreslet LB, Ingham AC, Iversen S, Lilje B, et al. 皮膚上のブドウ球菌群集は、アトピー性皮膚炎および疾患の重症度と関連する。Microorganisms. 2021 Feb;9(2):432. doi: 10.3390/microorganisms9020432. pmid:33669791
5.カレML. メタゲノミクスデータの統計解析. Genomics Inform. 2019;17(1):e6. doi: 10.5808/GI.2019.17.1.e6. pmid:30929407.
6.ウォーレンZD. 16Sアンプリコンシーケンスから得られた2つの大規模なパーキンソン病腸内細菌叢データセットを用いた、存在量差の検定方法の比較研究。BMC Bioinformatics. 2021 Dec 1;22(1):1-29. doi: 10.1186/s12859-021-04193-6. pmid:34034646.
7.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome Datasets Are Compositional: And This Is Not Optional. Front Microbiol. 2017 Nov;8:2224. doi: 10.3389/fmicb.2017.02224. pmid:29187837.
8.Gloor GB, Reid G. Compositional Analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol. 2016 Apr;62(8):692-703. doi: 10.1139/cjm-2015-0821. pmid:27314511
9.Gloor GB、Wu JR、Pawlowsky-Glahn V、Egozcue JJ. It's all relative: Analyzing microbiome data as compositions. Ann Epidemiol. 2016 May;26(5):322-9. doi: 10.1016/j.annepidem.2016.03.003. pmid:27143475.
10.Quinn TP, Erb I, Richardson MF, Crowley TM. シーケンスデータをコンポジションとして理解する:展望とレビュー。バイオインフォマティクス. 2018 Aug;34(16):2870-8. doi: 10.1093/bioinformatics/bty175. pmid:29608657.
11.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depends upon data characteristics. Microbiome. 2017 Mar;5(1):27. doi: 10.1186/s40168-017-0237-y. pmid:28253908
12.Aitchison J. The Statistical Analysis of Compositional Data. J R Stat Soc Ser B. 1982;44(2):139-77.
13.Lin H, Peddada S Das. 微生物組成の解析:正規化と差分存在量解析のレビュー(A review of normalization and differential abundance analysis). NPJ Biofilms Microbiomes. 2020 Dec;6(1):60. doi: 10.1038/s41522-020-00160-w. pmid:33268781
14.Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, et al. 基準フレームによる微生物組成測定基準の確立。Nat Commun. 2019 Jun;10(1):2719. doi: 10.1038/s41467-019-10656-5. pmid:31222023.
15.McMurdie PJ, Holmes S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLOS Comput Biol. 2014;10(4):e1003531. doi: 10.1371/journal.pcbi.1003531. pmid:24699258
16.Mallick H, Ma S, Franzosa EA, Vatanen T, Morgan XC, Huttenhower C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017 Nov 30;18(1):1-16. doi: 10.1186/s13059-017-1359-z. pmid:29187204.
17.Hawinkel S, Mattiello F, Bijnens L, Thas O. A broken promise: microbiome differential abundance methods do not control false discovery rate. Brief Bioinform. 2019 Jan;20(1):210-21. doi: 10.1093/bib/bbx104. pmid:28968702.
18.Jonsson V, Österlund T, Nerman O, Kristiansson E. 比較メタゲノミクスにおける差次量遺伝子の同定手法の統計的評価。BMC Genomics. 2016 Jan;17:78. doi: 10.1186/s12864-016-2386-y. pmid:26810311
19.Calgaro M, Romualdi C, Waldron L, Risso D, Vitulo N. Assessment of statistical methods from single cell, bulk RNA-seq, and metagenomics applied to microbiome data. Genome Biol. 2020 Aug;21(1):191. doi: 10.1186/s13059-020-02104-1. pmid:32746888.
20.Khomich M, Måge I, Rud I, Berget I. Analysing microbiome intervention design studies: 代替多変量統計手法の比較。PLoS One. 2021 Nov 1;16(11):e0259973. doi: 10.1371/journal.pone.0259973. pmid:34793531
21.Hawinkel S, Rayner JCW, Bijnens L, Thas O. シークエンスカウントデータは負の二項分布でうまく適合しない。PLoS One. 2020 Apr 1;15(4):e0224909. doi: 10.1371/journal.pone.0224909. pmid:32352970
22.Deek RA, Li H. マイクロバイオーム研究のためのゼロ膨張潜在ディリクレ配分モデル。Front Genet. 2021 Jan 22;11:1844. doi: 10.3389/fgene.2020.602594. pmid:33552122
23.Patuzzi I, Baruzzo G, Losasso C, Ricci A, Di Camillo B. metaSPARSim: a 16S rRNA gene sequencing count data simulator. BMC Bioinformatics. 2019 Nov;20(Suppl 9):416. doi: 10.1186/s12859-019-2882-6. pmid:31757204.
24.Fernandes AD、Reid JNS、Macklaim JM、McMurrough TA、Edgell DR、Glor GB. 高スループットシーケンスデータセットの解析の統一:組成データ解析によるRNA-seq、16S rRNA遺伝子シーケンスおよび選択的増殖実験の特徴づけ。Microbiome. 2014 May;2(1):15. doi: 10.1186/2049-2618-2-15. pmid:24910773.
25.Liu T, Zhao H, Wang T. An empirical Bayes approach to normalization and differential abundance testing for microbiome data. BMC Bioinformatics. 2020 Jun;21(1):255. doi: 10.1186/s12859-020-03552-z. pmid:32493208.
26.Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. マイクロバイオームの組成の分析:微生物の組成を研究するための新しい方法。Microb Ecol Heal Dis. 2015 May;26:27663. doi: 10.3402/mehd.v26.27663. pmid:26028277.
27.Lin H, Peddada S Das. バイアス補正によるマイクロバイオームの組成の解析。Nat Commun. 2020;11(1):3514. doi: 10.1038/s41467-020-17041-7. pmid:32665548.
28.Martin BD、Witten D、Willis AD. modeling microbial abundances and dysbiosis with beta-binomial regression. Ann Appl Stat. 2020 Mar;14(1):94-115. doi: 10.1214/19-aoas1283. pmid:32983313.
29.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, et al. Pop-scale meta-omics studiesにおける多変量関連発見。PLOS Comput Biol. 2021 Nov 1;17(11):e1009442. doi: 10.1371/journal.pcbi.1009442. pmid:34784344
30.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene survey. Nat Methods. 2013 Dec;10(12):1200-2. doi: 10.1038/nmeth.2658. pmid:24076764
31.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan;26(1):139-40. doi: 10.1093/bioinformatics/btp616. pmid:19910308
32.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014 Dec;15(12):550. doi: 10.1186/s13059-014-0550-8. pmid:25516281.
33.McMurdie PJ, Holmes S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One. 2013 Apr;8(4):e61217. doi: 10.1371/journal.pone.0061217. pmid:23630581
34.Holm S. A Simple Sequentially Rejective Multiple Test Procedure. Scand J Stat. 1979;6(2):65-70.
35.Clausen DS, Willis AD. マイクロバイオーム実験における複雑な測定誤差のモデリング。2022 Apr 27; https://arxiv.org/abs/2204.12733
36.McLaren MR, Willis AD, Callahan BJ. メタゲノムシーケンス実験における一貫性のある修正可能なバイアス。Elife. 2019 Sep 1;8. doi: 10.7554/eLife.46923. pmid:31502536.
37.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of healthy human microbiome. Nature. 2012 Jun;486(7402):207-14. doi: 10.1038/nature11234. pmid:22699609
38.Methé BA, Nelson KE, Pop M, Creasy HH, Giglio MG, Huttenhower C, et al. ヒトマイクロバイオーム研究のための枠組み. Nature. 2012 Jun;486(7402):215-21. doi: 10.1038/nature11209. pmid:22699610
39.He X, Parenti M, Grip T, Lönnerdal B, Timby N, Domellöf M, et al. 牛MFGM添加ミルクまたは標準ミルクを母乳で育った乳児を参照として与えた乳児の糞便マイクロバイオームとメタボローム:ランダム化比較試験。Sci Rep. 2019 Aug;9(1):11589. doi: 10.1038/s41598-019-47953-4. pmid:31406230.
40.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of gut microbial ecosystem in inflammatory bowel disease. Nature. 2019 May;569(7758):655-62. doi: 10.1038/s41586-019-1237-9. pmid:31142855.
41.Chen L, Reeve J, Zhang L, Huang S, Wang X, Chen J. GMPR: ゼロインフレートカウントデータのロバストな正規化法、マイクロバイオームシーケンスデータへの適用。PeerJ. 2018 Apr 2;2018(4):e4600. doi: 10.7717/peerj.4600. pmid:29629248.
42.Liu Y, Zhou J, White KP. RNA-seqによる差分発現研究:More sequence or more replication? Bioinformatics. 2014 Feb;30(3):301-4. doi: 10.1093/bioinformatics/btt688. pmid:24319002.
43.Baccarella A, Williams CR, Parrish JZ, Kim CC. サンプル数とリード深度がRNA-Seq解析ワークフローの性能に与える影響の経験的評価。BMC Bioinformatics. 2018 Nov;19(1):423. doi: 10.1186/s12859-018-2445-2. pmid:30428853.
44.Rong R, Jiang S, Xu L, Xiao G, Xie Y, Liu DJ, et al. MB-GAN: Generative Adversarial Network によるマイクロバイオームシミュレーション. Gigascience. 2021 Jan 29;10(2):1-11. doi: 10.1093/gigascience/giab005. pmid:33543271.
45.Ma S, Ren B, Mallick H, Moon YS, Schwager E, Maharjan S, et al. A statistical model for describing and simulating micro community profiles. PLOS Comput Biol. 2021 Sep 1;17(9):e1008913. doi: 10.1371/journal.pcbi.1008913. pmid:34516542
46.Goodfellow IJ, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, et al. Generative Adversarial Nets. Adv Neural Inf Process Syst. 2014;27.
47.Durazzi F, Sala C, Castellani G, Manfreda G, Remondini D, De Cesare A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci Reports 2021 111. 2021 Feb 4;11(1):1-10. doi: 10.1038/s41598-021-82726-y. pmid:33542369
48.Brumfield KD, Huq A, Colwell RR, Olds JL, Leddy MB. 公開されているNEONデータを用いた全ゲノムショットガンと16Sアンプリコンメタゲノムシーケンスの微生物分解能。PLoS One. 2020 Feb 1;15(2):e0228899. doi: 10.1371/journal.pone.0228899. pmid:32053657.
49.Lewis S, Nash A, Li Q, Ahn TH. 機械学習を用いた16Sと全ゲノム犬マイクロバイオームの比較。BioData Min. 2021 Dec 1;14(1):1-15. doi: 10.1186/s13040-021-00270-x. pmid:34419136
50.Cario MC, Nelson BL. 任意のマージナル分布と相関行列を持つランダムベクトルのモデリングと生成 (pp. 1-19). Technical Report, Department of Industrial Engineering and Management Sciences, Northwestern University, Evanston, Illinois. 1997.
51.Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Microbial Ecological NetworksのSparse and Compositionally Robust Inference of Microbial Ecological Networks. PLOS Comput Biol. 2015 May 1;11(5):e1004226. doi: 10.1371/journal.pcbi.1004226. pmid:25950956
52.Prost V, Gazut S, Brüls T. A zero inflated log-normal model for inference of sparse microbial association networks. PLOS Comput Biol. 2021 Jun 1;17(6):e1009089. doi: 10.1371/journal.pcbi.1009089. pmid:34143768
53.Baruzzo G, Patuzzi I, Di Camillo B. Beware to ignore the rare: How imputing zero-values can improve quality of 16S rRNA gene studies results. BMC Bioinformatics. 2021 Nov 1;22(15):1-34. doi: 10.1186/s12859-022-04587-0. pmid:35130833.
54.Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022 Dec 1;13(1). doi: 10.1038/s41467-022-28034-z. pmid:35039521
55.Morton JT, Sanders J, Quinn RA, McDonald D, Gonzalez A, Vázquez-Baeza Y, et al. Balance Trees Reveal Microbial Niche Differentiation. mSystems. 2017 Feb;2(1):e00162-16. doi: 10.1128/mSystems.00162-16. pmid:28144630
56.Rivera-Pinto J, Egozcue JJ, Pawlowsky-Glahn V, Paredes R, Noguera-Julian M, Calle ML. 天秤:マイクロバイオーム解析の新たな視点 mSystems. 2018 Jul;3(4):e00053-18. doi: 10.1128/mSystems.00053-18. pmid:30035234.
ViewFigures(ビューフィギュア) (14)
ViewReaderのコメント
見る著者について
ビューメトリクス
ViewMediaの取材
ViewPeer レビュー
記事をダウンロードする(pdf)
ダウンローディングクライテーション
メールで送る
PLOS JournalsPLOS Blogs
トップページへ戻るトップページへ戻る
会社概要
フルサイト
フィードバック
連絡先
個人情報保護方針
利用規約
メディアに関するお問い合わせ
PLOSは非営利の501(c)(3)法人で、#C2354500、米国カリフォルニア州サンフランシスコに拠点を置いています。
この記事が気に入ったらサポートをしてみませんか?