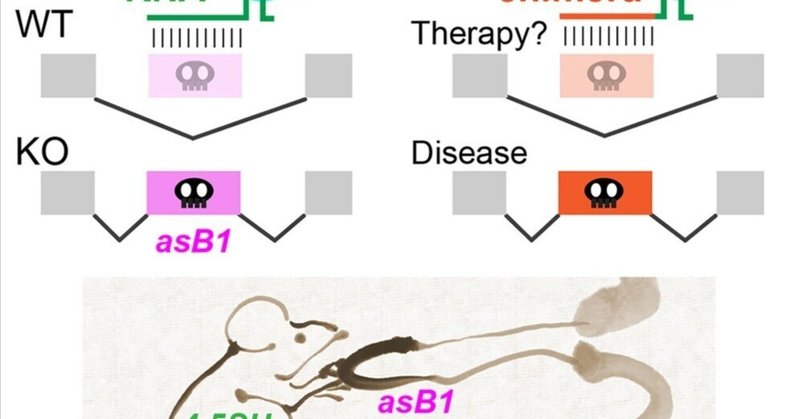
4.5SH RNAはマウスにおけるSINE B1の有害なエクソン化に対抗する

本文へスキップ記事へスキップ
エルゼビアロゴ
分子細胞
オンラインで入手可能 2023年12月13日
In Press, Corrected Proofこれは何ですか?
論文
4.5SH RNAはマウスにおけるSINE B1の有害なエクソン化に対抗する
https://www.sciencedirect.com/science/article/pii/S1097276523009644?dgcid=author

著者リンク オーバーレイパネルを開く吉本怜1、中山雄太2、野村郁子2、山本郁子2、中川夢佳1、田中茂幸1、栗原美鈴2、鈴木悠3、 小林武彦 3、小塚畑浩子 4、大山正明 4、水戸真理 5、岩崎慎太郎 5 6、山崎智弘 7、廣瀬哲朗 7 8、荒木公美 9 10、中川真一 2 11
もっと見る
概要
シェア
引用
https://doi.org/10.1016/j.molcel.2023.11.019
権利とコンテンツを入手する
ハイライト
4.5SH KO細胞はレトロトランスポゾンSINE B1挿入による異常なエクソナイゼーションを示す
4.5SH RNAは異常エクソンと塩基対を形成し、エクソンスキッピングのための因子をリクルートする。
4.5SHキメラRNAはプログラム可能なスプライシング制御因子として機能する。
4.5SH RNAはマウスの胚生存に重要である。
まとめ
4.5SH RNAは、プレmRNAスプライシング制御因子が濃縮された核の斑点に局在する、高濃度の齧歯類特異的な低分子ノンコーディングRNAである。4.5SH RNAの生理学的機能を調べるため、我々は4.5SH RNAの発現を欠損した変異マウスを作製した。この変異マウスは胚致死を示したことから、4.5SH RNAはマウスに必須の種特異的ノンコーディングRNAであることが示唆された。RNAシークエンシング解析の結果、4.5SH RNAはレトロトランスポゾンSINE B1(asB1)のアンチセンス挿入による異常なエクソナイゼーションからトランスクリプトームを保護していることが明らかになった。機構的には、4.5SH RNAは標的認識領域を介して相補的なasB1を含むエクソンと塩基対を形成し、5′ステムループ領域を介してHnrnpmを含むエフェクタータンパク質をリクルートする。このような4.5SH RNAのモジュール構造により、目的のエクソンのスキップを誘導するプログラム可能なスプライシング制御因子を設計することができる。この結果はまた、スプライシング制御ノンコーディングRNAの一般的な存在を示唆している。
グラフィカル抄録
ダウンロード 高解像度画像ダウンロード(186KB)
ダウンロード フルサイズ画像のダウンロード
キーワード
4.5SH RNAノンコーディングRNAエキソンスキッピング代替スプライシングレトロトランスポゾンSINE B1Hnrnpm
はじめに
レトロトランスポゾンは高等真核生物のゲノムのかなりの部分を構成している。特に、7SL由来のレトロトランスポゾン、すなわちげっ歯類のSINE B1と霊長類のAluは、これらの種の中で最も普及している可動要素である。1アンチセンス方向に挿入された場合、SINE B1ファミリーのレトロトランスポゾンは、ポリピリミジントラクト、ほぼ最適な3′スプライス部位、および最適でない5′ドナー部位を含む、分岐点配列を除くエクソンの定義に必要な全ての重要な配列要素を提供する2。このように、イントロンへのSINE B1ファミリーレトロトランスポゾンのアンチセンス挿入は、分岐点コンセンサス配列の下流に挿入された場合、早発停止コドンやフレームシフト変異を導入し、次いでスプライス部位を最適化する変異を導入する、有害なエクソンの強力な供給源となる2,3。ヒト細胞は、Aluのポリピリミジントラクト配列に対してU2AF2と競合するHNRNPCを用いて、SINE B1ファミリーレトロトランスポゾンのヒトでの対応物であるAluエレメントのこのような有害なエクソン化に対抗することが示されている。
膨大な数のノンコーディングRNAが高等真核生物のゲノムから転写されており、その多くは種特異的で核内に局在している5,6。4.5SH RNAは、1970年代に同定された90塩基の典型的な核内ノンコーディングRNAであり7,8,9、マウスやラットなどの小型で短命なげっ歯類に特異的に見出される10。4.5SH RNAの塩基配列はSINE B1と非常に相同性が高いが、4.5SHにはSINEのレトロトランスポジションに必要なAリッチテールがないため、トランスポーズ活性がない1。4.5SH遺伝子はマルチコピー遺伝子であり、タンデムリピートの大きなクラスターを形成し、細胞あたり10,000分子以上に達する非常に豊富な転写産物を産生する11。4.5SH RNAは様々な組織や細胞種でユビキタスに発現し、転写産物はスプライシング因子に富む核の斑点に局在する12。
本研究では、4.5SH RNAの発現を欠損させた変異マウスを作製し、このマウスが初期胚致死を示すことを見出した。注目すべきことに、変異細胞の転写産物には数百の新規エクソンが異常に含まれており、その大部分はSINE B1のアンチセンス挿入を含んでいた。ミニ遺伝子解析の結果、4.5SH RNAはSINE B1を含む破壊的エクソンと塩基対を形成し、3′スプライス部位の利用を阻害することによって、そのエクソンの取り込みを阻害することが明らかになった。標的部位への4.5SH RNAの塩基対形成はエクソンスキッピング活性には十分ではなく、4.5SH RNAの5′に位置するステムループ構造に結合するHnrnpmを含むエフェクタータンパク質のリクルートが必要であった。4.5SH RNAのモジュール構造により、標的認識モジュールを標的エクソンに相補的な配列に置き換えることで、目的のエクソンのスキップを誘導できるプログラム可能なスプライシング制御因子を作り出すことができた。この結果はまた、Hnrnpmと会合し、相補的配列を含む標的エクソンの代替スプライシングを制御するノンコーディングRNAの一般的な存在を示唆している。
結果
4.5SH RNAはマウスの初期発生とES細胞の増殖に必須である
4.5SH RNAの生理学的機能を調べるため、このマルチコピー遺伝子の変異マウスを作成した。まず、パルスフィールドゲル電気泳動を行い、4.5SH遺伝子クラスターのサイズとコピー数を正確に推定した。この遺伝子クラスターは、最新のマウスゲノム解析でもこのゲノム領域に大きなギャップがあり、あいまいなままであった(図1A)。4.5SHを含むリピートユニット内を切断する制限酵素でゲノムDNAを処理すると、既述のように、サザンブロット上で均一な長さのバンド(4.2kb)が得られた11(図1B)。対照的に、遺伝子クラスターの外側を切断する酵素では、約900kbの単一バンドが得られた(図1B)。このことは、4.5SHの200コピー以上が、4.5SH遺伝子の全てではないにしても大部分を含むこの単一遺伝子座で、大きなタンデム型クラスターを形成していることを示唆している。メガベースサイズの大きな4.5SHクラスターを削除するため、相同組換え効率を高めるために、CRISPR-Cas9を用いて遺伝子標的ベクターのアームの近位領域を切断した(図1AおよびS1A-S1D)。我々は、4.5SHクラスター全体を欠損させた胚性幹(ES)細胞クローンを得ることに成功し、これを用いて4.5SHノックアウト(KO)マウスを作製した(図S1E-S1H)。E2.5-E3.5といった着床前段階のKO胚には、明らかな異常は見られなかった(図1Cおよび1D)。しかし、E9.5より後期ではホモ接合体を得ることができず(図1C)、E6.5では異常な細胞凝集体のみが回収された(図1E)。4.5SH RNAの細胞機能と分子機能をさらに調べるため、胚盤胞期胚の内部細胞塊から誘導される多能性細胞である胚性幹(ES)細胞を、野生型(WT)とKO胚から樹立した(図1F)。4.5SHのKO ES細胞では、アルカリホスファターゼやNanogなどの多能性マーカーの発現は維持されていたが(図1G)、WT ES細胞と比較して細胞増殖の低下が観察された(図1H)。外因性4.5SHの発現は細胞増殖の低下を救った(図1Fと1H)ことから、クラスター内のcis-DNAエレメントではなく、転写された4.5SH RNAがES細胞の増殖を制御していることが示唆された。
ダウンロード 高解像度画像ダウンロード(1MB)
ダウンロード フルサイズ画像のダウンロード
図1. 4.5SH KOマウスは胚致死を示す
(A)6番染色体上の4.5SHクラスターのゲノム構成と、4.5SH KOマウスを作製するためのターゲティング戦略の概略図。
(B)従来法(左)とパルスフィールドゲル電気泳動法(右)を用いた4.5SHのサザンブロット解析。
(C)ヘテロ接合体(+/-)マウスを交配して得られた子孫の遺伝子型。遺伝子型判定はin situハイブリダイゼーションで検出された4.5SH RNAの発現で判断したため、E2.5-E3.5におけるヘテロ接合体胚と野生型胚を明確に区別することはできなかった。
(D)8細胞期のWTおよび4.5SH KO胚のノマルスキーDIC画像に、LNAプローブ(緑)で検出した4.5SH RNAシグナルを重ねたもの。
(E)WTおよび4.5SH KOマウスのE6.5期胚の横断面を、4.5SHプローブ(緑)とDAPI(マゼンタ)を用いて低倍率(上段)と高倍率(下段)で可視化したもの。上段の点線は胚組織の輪郭を示す。下段の点線は胚組織の細胞核を示す。アスタリスクは、複数の非励起チャンネルで検出されたシグナルから判断した、細胞質内の非特異的自家蛍光シグナルを示す。
(F)WT、KO、およびレスキュー(res)ES細胞における4.5SH発現のRT-qPCR解析。
(G)WT、4.5SH KO、およびレスキュー(res)ES細胞におけるアルカリホスファターゼ活性(AP)とNanogの発現。
(H)WT、4.5SH KO、およびレスキュー(res)ES細胞の増殖。エラーバーは生物学的二重記録からの標準偏差を表す。スケールバー、(E)の上下のパネルではそれぞれ100μmと20μm、(G)の上下のパネルではそれぞれ200μmと10μm。
4.5SH KO ES細胞では、SINE B1のアンチセンス挿入を含む破壊的なクリプトエキソンが異常に含まれていた。
先に述べたように12、4.5SH RNAは、ES細胞におけるSrsf1の発現によって同定された核の斑点に局在した(図2A)。核の斑点はスプライシング因子に富んでいることから13、4.5SH RNAがプレmRNAのスプライシングを制御していると予想された。4.5SH KO ES細胞で変化したスプライシングイベントを調べるため、ショートリードのディープシーケンスデータから代替スプライシングイベントの変化を検出できるrMATS解析を行った14。WTおよび4.5SH KO ES細胞から得られたRNAシーケンスデータを用いて、WTとKO細胞の間でスプライシングが異なる160のカセットエキソン(偽発見率[FDR]<0.001)を検出し、そのうち100(63%)のエキソンがKO細胞で優先的に見つかった(図2B;表S1)。注目すべきことに、KOで濃縮されたエクソンの75%はRefSeq-unannotated cryptic exonであり(図2B; 表S1)、これはES細胞の正常なトランスクリプトームでは見られなかった。
ダウンロード 高解像度画像ダウンロード(1MB)
ダウンロード フルサイズ画像のダウンロード
図2. 4.5SH KO ES細胞では劇症エクソンが異常に含まれている。
(A)ES細胞における4.5SH RNA(緑)と核スペックルマーカーSrsf1(マゼンタ)の同時検出。
(B)RefSeqで注釈付けされたエクソン(青)と注釈付けされていない新規エクソン(マゼンタ)のボルケーノプロットと、KOで濃縮されたエクソンの概略図。FDR=0のエクソンは、ボルケーノプロットの同じ無限(∞)Y値上に外れ値として示されている。
(C) MEME解析によって明らかになったKO特異的クリプティックエクソンに濃縮されたモチーフ。
(D)コンセンサスモチーフとasB1および4.5SH RNAとの相対位置。
(E)特定のエクソンにおけるインクルージョンの違いを示すボルケーノプロット。マゼンタの点はレトロトランスポゾンが挿入されたエクソンを示す:sB1とasB1はそれぞれSINE B1のセンス挿入とアンチセンス挿入、sB2とasB2はそれぞれSINE B2のセンス挿入とアンチセンス挿入。FDR=0のエクソンは、ボルケーノプロットの同じ無限(∞)Y値上に外れ値として示されている。
(F)マップされた配列リードと遺伝子アノテーションのゲノムブラウザビュー。マゼンタのボックスはKO特異的RefSeq注釈なしクリプティックエクソンを示し、青いバーはasB1の位置を示す。
(G)2つの独立したWT、KO、およびレスキュー(res)ES細胞を用いたRT-PCR産物のキャピラリー電気泳動のゲル状図。
(H)(G)に示したデータの定量。各ドットは各ES細胞株におけるエクソンスキップ転写産物の割合を示す。
(I) WT、KO、およびレスキュー(res)ES細胞におけるSmg6とSmchd1のウェスタンブロット解析。数字はバンドの相対強度の定量化を示す。Smg6とSmchd1については、Gapdhの同じローディングコントロール画像を用いた。スケールバーは5μm。
次に、これらのKOで濃縮されたエクソンに共通する特徴的な配列があるかどうかを調べた。RefSeqエクソンと比較すると、これらのエクソンは典型的なエクソンを思わせる特徴を示したが(図S2A-S2E)、MEME15を用いたモチーフ解析により、レトロトランスポゾンSINE B1(asB1)のアンチセンス挿入部と非常に相同性の高い3つのコンセンサス配列が明らかになり、そのうちの2つ(モチーフ1と3)は4.5SHと相補的であった(図2C、2D、S2F)。KOで濃縮されたエクソンにおけるasB1の優先的な出現は鎖特異的であり、SINE B2のような他のレトロトランスポゾンはこれらのエクソンでは濃縮されなかった(図2E)。実際、asB1は、上流のポリピリミジントラクト、ピリミジンとGで挟まれた3′スプライス部位の配列AG、GTを含む5′スプライス部位など、エクソンの定義に必要な配列モチーフを生来含んでいる。asB1を含むクリプティックエキソンのうち、81%は宿主遺伝子にPTC変異またはフレームシフト変異を導入しており(図S2G)、4.5SH KOマウスの胚致死の主な原因は、asB1を含む有害なエキソンの挿入であることが示唆された。
asB1含有エクソンのKO特異的封入をさらに確認するために、WTおよび4.5SH KO ES細胞由来のRNAを用いて、Slc25a40、Smchd1、Smg6を含む代表的な宿主遺伝子の逆転写酵素ポリメラーゼ連鎖反応(RT-PCR)を行った(図2F-2H)。重要なことは、これらの遺伝子のasB1を含むクリプトエキソンはほとんどスキップされ、WT細胞では検出されなかったことである(図2Gおよび2H)。しかしKO細胞では、これらの異常エクソンを含む転写産物が優勢になり、外因性4.5SH RNAを発現しているレスキュー細胞ではもはや観察されなかった(図2Gと2H)。代表的遺伝子のこれらのエクソンには早発停止コドンが含まれており、Smchd1とSmg6の全長タンパク質産物の減少はウェスタンブロットで確認された(図2I)。代表的な宿主遺伝子のタンパク質発現低下は、4.5SHの外因性導入により回復した(図2I)。4.5SH KO ES細胞における代表的な遺伝子のasB1含有エクソンの包含は、核内RNAと細胞質RNAの両方で一貫して観察された(図S3)ことから、asB1包含の上昇は、転写産物の細胞内局在の違いによるものではないことが示唆された。さらに、これらの所見は、RT反応に用いるプライマーがオリゴdTであろうとランダムプライマーであろうと一貫しており(図S3)、4.5SH KO ES細胞のasB1含有エクソンは成熟mRNA中に存在することが示された。
次に、差次発現遺伝子の解析を行ったところ、Ensembl注釈付き遺伝子のうち1,014個が、4.5SH KO ES細胞において有意に変化した発現パターン(絶対対数2倍変化>1、FDR<0.01)を示した(図S4A)。これらには、リボソーム生合成、脂肪酸分解、スプライソソーム構成要素などに関連する遺伝子が含まれる(図S4BおよびS4C)。これらの所見は、asB1エクソンの封入が多数の宿主遺伝子の機能を擾乱し、様々な細胞プロセスへの影響の連鎖を引き起こし、4.5SH KO ES細胞における細胞増殖の低下をもたらすことを示唆している。注目すべきは、Cul1、Ezh2、Pdia4、Zfp786を含む4.5SH遺伝子クラスターに隣接する遺伝子が、すべて微妙なアップレギュレーション(log2-fold change = 0.32〜0.72)を示したことである(図S4DとS4Eの緑色の点)。このことは、4.5SHクラスターの欠失がこの遺伝子座の高次クロマチン構成に影響を与え、遺伝子発現に影響を与えた可能性を示唆している。また、asB1を含むエクソンがKO特異的にインクルージョンしている遺伝子は、ダウンレギュレーションされる傾向があることもわかった(図S4Dのマゼンタの点)。このダウンレギュレーションは、asB1含有エクソン封入体から早発停止コドンおよび/またはフレームシフト変異が導入され、ナンセンス媒介崩壊経路を通じてこれらの異常転写産物が分解されるためと考えられる16。
4.5SH RNAは塩基対形成を介して標的エクソンを認識し、3′スプライス部位の使用を阻害する
4.5SH RNAの分子機構をさらに探るため、我々はasB1を含むSlc25a40の暗号エクソン(asB1-Slc25a40)を、そのフランキング配列とともにアデノウイルス由来のよく知られたスプライシングレポーター17のイントロンに挿入し、ミニ遺伝子を構築した(図3A)。このミニ遺伝子を4.5SH RNAを発現していないヒト由来のHEK293細胞に導入したところ、asB1-Slc25a40はほぼ完全にスプライシングされた(図3B)。このことは、4.5SHが発現していない細胞では、暗号エクソンが真正のエクソンとして認識されていることを示唆している。対照的に、4.5SH RNAを内因性に発現するマウス細胞株Neuro2Aにミニ遺伝子を導入すると、asB1-Slc25a40の55%がスキップされ、短い産物が生じた(図3B)。また、HEK293細胞でU6プロモーターの制御下に4.5SH RNAを発現させた場合にも、スキップされた産物が観察された(図3B)。このことから、ミニ遺伝子システムを用いて、asB1の4.5SH RNA依存的エキソンスキッピングを再現することに成功したことが示された。
ダウンロード 高解像度画像ダウンロード(595KB)
ダウンロード フルサイズ画像のダウンロード
図3. 4.5SH RNAは標的asB1含有エクソンと塩基対を形成し、3′スプライス部位の使用を阻害する。
(A)Slc25a40の暗号エクソン(asB1-Slc25a40)とU6プロモーター(U6 pro)で駆動される4.5SH発現ベクターを含むミニ遺伝子の概略図。
(B)asB1-Slc25a40代替スプライシングのキャピラリー電気泳動、4.5SH RNA発現のノーザンブロット解析、およびスキップ産物の割合の定量化のゲル状図。アスタリスクはバイオアナライザーに表示されたヘテロ二重鎖バンドの位置を示す。
(C) (D)で使用した互い違いの突然変異。
(D)野生型または変異型4.5SH/asB1-Slc25a40を発現するミニ遺伝子導入HEK293細胞におけるasB1-Slc25a40の代替スプライシングとスキップ産物の定量。アスタリスクはバイオアナライザーに表示されるヘテロ二重鎖バンドの位置を示す。
(E)asB1-Slc25a40の3′または5′スプライス部位(SS)を含むスプリットミニゲンの概略図。
(F) 4.5SH RNAの発現に伴う、スプリットミニゲンをトランスフェクトしたHEK293細胞におけるasB1-Slc25a40のスプライシング。(B)、(D)、(F)のエラーバーは、生物学的3連プレートからの標準偏差を表す。
次に、相補的変異を用いて、4.5SH RNAが塩基対形成を介して標的エクソンに結合するかどうかを調べた(図3C)。塩基対形成を阻害する変異をasB1-Slc25a40または4.5SH RNAのいずれかに導入すると、標的エクソンのスキッピング活性が低下した(図3D)。塩基対形成をレスキューする相互変異を同時に導入すると、スキップ活性の低下は完全に回復した(図3D)。このことは、4.5SH RNAが標的エクソンにハイブリダイズして、そのインクルージョンを阻害していることを示唆している。スプリットミニゲインを用いて、4.5SH RNAがasB1-Slc25a40の5′スプライス部位ではなく、3′スプライス部位の使用を阻害することも見いだした(図3Eおよび3F)。
SL1-4.5SHは4.5SH RNAのエキソンスキッピング活性に必要である。
次に、4.5SH RNAの機能に必要な配列要素を、4ヌクレオチドが重複しない欠失を持つ一連の欠失変異体を用いて同定することを試みた(図4Aおよび4B)。asB1-Slc25a40スキップの顕著な減少につながる欠失は、RNAfold2によって予測された3つのステムループ領域(SL1、SL2、SL3)のうちの1つにマップされた18。ノーザンブロット解析の結果、SL2またはSL3の後半の欠失は変異型4.5SH RNAの発現を減少させたが、SL1の欠失は外来発現RNAの量に影響を与えずにエクソンスキッピング活性に特異的に影響を与えた(図4Cおよび4D)。このことから、4.5SH RNAのSL1(SL1-4.5SH)は4.5SH RNAのエクソンスキッピング活性に必須であることが示唆された。次に、SL1-4.5SHは、Ppil4やEya3など、SL1-4.5SHと塩基対を形成するモチーフ3を持たない特定の宿主遺伝子の標的エクソンと塩基対を形成しないことに気づいた(図4Bと4E)。さらに、MEMEで同定されたモチーフ3は、KOで濃縮されたasB1を含むエクソンの41%にしか見られなかった(図S5AおよびS5B)。これらの観察結果は、SL1-4.5SHがハイブリダイゼーションに基づく標的認識以外のユニークな機能を持つことを示唆している。この可能性を検証するために、SL1-4.5SHを、相同配列を持ちながら異なる予測二次構造を示すSINE B1の対応する領域(SL1-B1)に置き換えた(図4F)。WTの4.5SH RNAとは異なり、SL1-B1とのキメラ分子は、標的配列とより安定な二次構造を形成すると予測されるにもかかわらず、asB1-Slc25a40の効率的なエクソンスキッピングを誘導できなかった(図4Gおよび4H)(図4I)。これらの観察から、標的エクソンへの塩基対形成は、それだけではエクソンスキッピングを誘導するには不十分であることが示唆される。その代わりに、4.5SH RNAはasB1と相補的な標的認識モジュールと潜在的なエフェクター結合モジュールであるSL1-4.5SHの2つのモジュールから構成されていると予測された(図4B)。この考えは、標的認識モジュールの相互変異が変異asB1-Slc25a40のエクソンスキッピングを回復させた前述の結果と一致していた(図3Cおよび4B)。そこで我々は、4.5SH RNAと標的エクソンとの間の相補性を高めることによって、エクソンスキッピング効率を向上させることができるかどうかに興味を持った。そこで我々は、4.5SH RNAの標的認識モジュールを、asB1-Slc25a40と完全に塩基対になる配列に変更した。予想外なことに、完全にマッチした4.5SH RNAのエキソンスキッピング活性は、WTの4.5SH RNAに比べてむしろ低下しており(図4J)、4.5SH RNA依存性のエキソンスキッピングには単純なハイブリダイゼーションだけでは不十分であるという考えをさらに裏付ける結果となった。
ダウンロード 高解像度画像ダウンロード(1MB)
ダウンロード フルサイズ画像のダウンロード
図4. エフェクター結合モジュールと標的認識モジュールからなる4.5SH RNAのモジュール構造
(A)4.5SH RNAの予測二次構造。
(B) 4.5SH RNAのモジュール構造と欠失変異体の領域の模式図。マゼンタのバーは、標的asB1を含むエクソンのMEMEで同定されたモチーフ1とモチーフ3に相補的な領域を示す。緑色のバーはステムループ構造の領域を示す。シアンのバーは、図3Cに示した相互変異体においてヌクレオチドが置換されている領域を示す。
(C)ミニ遺伝子導入細胞で調べた、4.5SH欠失変異体の発現と、4.5SH RNAのエクソンスキッピング活性に対する欠失変異の影響のノーザンブロット解析。
(D) (C)の結果の要約。黄色の丸は4.5SH RNAの残基を表し、その欠失によりエクソンスキッピング活性が低下(50%未満)した。マゼンタの文字は、欠失によって発現のダウンレギュレーションが生じた残基を表す。
(E) 4.5SH RNAとPpil4およびEya2のモチーフ3を欠くクリプティックエキソンとの間に形成される予測二次構造。
(F)4.5SHの5′ステムループ(SL1-4.5SH)とSINE B1(SL1-B1)の配列と予測される二次構造。
(G)Slc25a40の暗号エクソン(asB1-Slc25a40)、U6駆動4.5SH RNAおよびキメラRNA含有SL1-B1を含むミニ遺伝子の概略図。
(H) 4.5SH RNAまたはSL1-B1-4.5SHを発現するミニ遺伝子導入HEK293細胞におけるasB1-Slc25a40の代替スプライシング。アスタリスクはバイオアナライザーに現れるヘテロ二重鎖バンドの位置を示す。
(I) asB1-Slc25a40と4.5SH RNA/SL1-B1-4.5SHの間に形成される二次構造の予測。SL1-B1-4.5SHは、エキソンスキッピング活性が低下しているにもかかわらず、野生型4.5SH RNAと比較して高い相補性を示している。
(J)野生型または完全一致(pm)4.5SH RNAをミニ遺伝子導入したHEK293細胞におけるasB1-Slc25a40の代替スプライシング。アスタリスクは、バイオアナライザーに現れるヘテロ二重鎖バンドの位置を表す。エラーバーは、(C)では生物学的二重標本、(H)と(J)では生物学的三重標本からの標準偏差を表す。
4.5SH RNAのSL1はエフェクタータンパク質と会合してエクソンスキッピングを誘導する
我々は、SL1-4.5SHがエクソンスキッピングを誘導するタンパク質を、B1を含むクリプティックエキソンとして標的にリクルートすると推論した。SL1-4.5SHと会合するエフェクタータンパク質を同定するために、ビオチン化SL1-4.5SHとマウス細胞株Neuro2Aから調製した核溶解物を用いてRNAプルダウン実験を行った(図5A)。複数のタンパク質が、SL1-B1よりもSL1-4.5SHに優先的に結合し、75 kDaのタンパク質は、1M NaClによるストリンジェントな高塩洗浄後も結合したままであった(図5Bおよび5C)。質量分析とその後のウェスタンブロット解析により、少なくとも3種類のRNA結合タンパク質、Hnrnpm、Sfpq、NonoがSL1-4.5SHに優先的に結合していることが明らかになった(図5Dと5E)。これらのRNA結合タンパク質のうち、Hnrnpmは1M NaCl存在下でもSL1-4.5SHに結合したままであった。このことは、SL1-4.5SHの遠位領域に位置する、以前の研究19で同定された最も代表的な結合モチーフGUGGUGGの存在と一致していた(図5F)。細胞内でこれらのRNA結合タンパク質が4.5SH RNAに結合していることをさらに確認するために、4.5SH RNAを発現しているHEK293細胞と非機能性のSL1-B1-4.5SH RNAを発現しているコントロール細胞を用いてCLIP解析を行った。免疫沈降したRNAをノーザンブロットで検出したところ、3つのタンパク質はすべて4.5SH RNAに結合したが、非機能性キメラ分子には結合しなかった(図5G)。
ダウンロード 高解像度画像ダウンロード(2MB)
ダウンロード フルサイズ画像のダウンロード
図5. 4.5SH RNAの5′ステムループはエフェクタータンパク質をリクルートして標的エクソンのインクルージョンを阻害する。
(A)エフェクタータンパク質を同定するための実験デザインの概略図。
(BおよびC)ビオチン化したSINE B1の5′ステムループ1(SL1-B1)および4.5SHの5′ステムループ1(SL1-4.5SH)でプルダウンしたSDS-PAGE分離タンパク質を、通常の塩濃度(150mM NaCl)下(B)および1M NaClで高塩洗浄した後(C)で銀染色した。
(D) SL1-B1とSL1-4.5SHで精製したタンパク質のLC-MS分析結果の散布図(各n =1)。各ドットは同定されたタンパク質を示す。PSM:ペプチド・スペクトル一致スコア。
(E)ウェスタンブロットで検出されたHnrnpm、Sfpq、NonoとSL1-4.5SHとの優先的会合。75kDaのHnrnpmは1M NaClでストリンジェントに洗浄してもSL1-4.5SHに結合したままである。
(F)SL1-4.5SHにおける以前に同定されたコンセンサスHnrnpm結合配列の存在。サンゴ色は以前の研究で同定された推定Hnrnpm結合部位を示す19。
(G) 4.5SH RNAのCLIP解析。FLAGタグのついたHnrnpm、Nono、SfpqをHEK293細胞で野生型4.5SH RNAまたはキメラSL1-B1-4.5SHと共発現させ、抗FLAG抗体を用いて免疫沈降させた。免疫沈降した4.5SHはノーザンブロット解析で検出した。これらのRNA結合タンパク質はSL1-B1-4.5SHではなく4.5SH RNAとUV照射依存的に結合している。
(H)NIH3T3細胞における4.5SH RNA(緑)とエフェクタータンパク質(マゼンタ)Hnrnpm、Sfpq、Nonoの同時検出とカラープロファイルプロット。黄色の線はカラープロファイルプロットで示された領域を表す。矢印は重なり合ったピークの位置を示す。スケールバー、5μm。
次に、これらのRNA結合タンパク質の核内分布を調べ、ES細胞を用いて4.5SH RNAのシグナルと比較した(図5H)。Hnrnpmはこの細胞型では1つか2つの大きな病巣を形成し(図5H、白矢印)、4.5SH RNAのシグナルとは重ならなかった。しかし、4.5SH RNAのピークシグナルの多くは、核形質内の他の領域におけるHnrnpmの局所的ピークシグナルと重なっていた(図5H、黒矢印)。SfpqとNonoのシグナルは、4.5SH RNAのシグナルとほぼ重なっていた(図5H、黒矢印)。ES細胞はNeat1_2を発現しておらず、そのためパラスペックルの形成が見られないので21、パラスペックルが目立つNIH3T3細胞で4.5SH RNAと他の会合タンパク質の発現も調べた。ES細胞と同様、4.5SH RNAのシグナルはほとんどがSrsf1のシグナルと重なり(図5H、矢印)、NIH3T3細胞でも主に核の斑点に局在していることが示された。Hnrnpmのシグナルも4.5SH RNAのシグナルに近接して検出されたが、重なる頻度はSrsf1に比べて減少していた(図5H、矢頭)。SfpqとNonoは主に傍棘に局在し、RNAとの同時検出に用いた実験条件では、核形質には弱いシグナルしか検出されなかった(図5H)。しかし、4.5SH RNAのピークシグナルの一部は、核形質におけるSfpqとNonoの局所的ピークシグナルと重なっていた(図5H、矢頭)。これらの観察から、4.5SH RNAとこれらのエフェクタータンパク質との結合は厳密に安定しているわけではなく、核内で動的に制御されていることが示唆される。
エフェクタータンパク質の機能的関与を調べるため、ノックダウン実験を行った。HNRNPMおよびSFPQを欠損させたHEK293細胞にミニ遺伝子を導入すると、asB1-Slc25a40のスキッピングが減少した(図6AおよびS6A)。このことは、これら2つのタンパク質が4.5SH RNAと機能的複合体を形成し、標的エクソンのスキッピングを誘導していることを示唆している。また、NONOで枯渇させた細胞では、統計的に有意ではなかったが、スキッピング活性のわずかな低下が観察された(図6A)。これは、NONOのノックダウンが非効率的であったためか(図S6A)、あるいはSfpqと相同性の高いNONOの冗長な機能のためかもしれない22。次に、MS2システムを用いて標的エクソンにエフェクタータンパク質をリクルートすることにより、エフェクタータンパク質の優性効果を調べた23(図6BおよびS6B)。予想通り、HNRNPM、SFPQ、NONOを強制的にリクルートすると、4.5SH RNA非存在下でasB1を含むエクソンのスキッピングが誘導されたが、MS2コートタンパク質をリクルートしてもほとんど効果はなかった(図6B)。エフェクタータンパク質の支配的な効果は、タンパク質が下流のイントロンにリクルートされた場合には観察されなかった(図6C)。これらの観察から、4.5SH RNAは標的認識モジュールを介して標的asB1を含むエクソンと塩基対を形成し、Hnrnpmや他の会合タンパク質を含むエフェクタータンパク質複合体をリクルートし、標的エクソンのスキップを誘導することが示唆される。
ダウンロード 高解像度画像ダウンロード(1MB)
ダウンロード フルサイズ画像のダウンロード
図6. 4.5SH RNA依存性エクソンスキッピングにおけるSL1-4.5SH関連エフェクタータンパク質の機能的関与
(A)HNRNPM、SFPQ、NONOを欠損させたHEK293細胞をミニジーントランスフェクトしたasB1-Slc25a40の代替スプライシング。HNRNPMとSFPQをノックダウンすると、エキソンスキッピングが減少した。大きなアスタリスクはコントロールと比較して統計的に有意な変化を示し(p < 0.016、Bonferroni補正後のWelchのt検定)、小さなアスタリスクはバイオアナライザーに現れるヘテロ二重鎖バンドの位置を示す。
(BおよびC)MS2システムを用いたミニ遺伝子のエクソン(B)および下流のイントロン(C)へのRNA結合タンパク質のリクルート、およびMS2コートタンパク質(MS2CP)に融合したRNA結合タンパク質を発現するHEK293細胞におけるasB1-Slc25a40の代替スプライシングの模式図。アスタリスクはコントロールと比較して統計的に有意な変化を示す(p < 0.0125、Bonferroni補正後のWelchのt検定)。
(D)ミニ遺伝子をトランスフェクトしたHNRNPC欠失HEK293細胞におけるasB1-Slc25a40の代替スプライシングと、ノックダウン(KD)によりHNRNPCが減少したことを確認するウェスタンブロット解析。注目すべきは、4.5SH RNAを介したasB1-Slc25a40のエキソンスキッピングは、HNRNPCの減少でも持続したことである。一方、HNRNPCの枯渇は、以前の研究で明らかにされたように、HELLSとKIAA1432からのasAluを含む内因性クリプティックエキソンのインクルージョンをもたらした4。
(E)Hnrnpcで枯渇させたマウスNeuro2A細胞における内因性asB1含有エクソンの代替スプライシングと、ノックダウン(KD)によりHnrnpcが減少したことを確認するウェスタンブロット解析。asB1を含むエクソンのスプライシングは影響を受けなかったが、CD55ミニ遺伝子からのasAluを含むエクソンのスキップはHnrnpcの枯渇によって減少した。(A)-(E)のエラーバーは生物学的3連データからの標準偏差を示す。
HNRNPCはU2AF2と競合することにより、ヒトのSINE B1ファミリーレトロトランスポゾンであるAluエレメントのアンチセンス挿入の異常なエクソン化を防ぐことが以前に示されている4。HNRNPCが4.5SH RNAのエクソンスキッピング活性にも必要であるかどうかを調べるために、HNRNPCを欠失させたHEK293細胞にスプライシングレポーターミニジーンと4.5SH発現ベクターを導入した。以前に報告されたように4、HELLSとKIAA1432のasAluを含むクリプトエキソンは、HNRNPCノックダウンにより異常スプライシングされた(図6D)。しかしながら、asB1-Slc25a40の4.5SH RNA依存性エキソンスキッピングは、HNRNPC非存在下でも影響を受けなかった(図6D)。asB1-Slc25a40およびasB1-Smg6のようなasB1を含むエクソンのスキッピングも、Hnrnpcで枯渇させたNeuro2A細胞では影響を受けなかった(図6E)。一方、CD55ミニ遺伝子からのasAluを含むエクソンのHnrnpc介在性スキッピングは、ネズミ細胞で顕著であった(図6E)。これらの結果は、小型のげっ歯類と霊長類が、それぞれノンコーディングRNAとRNA結合タンパク質を用いて、SINE B1ファミリーレトロトランスポゾンのアンチセンス挿入によるエクソン化の悪影響を軽減する戦略を独自に開発したことを示している。
4.5SH RNAのモジュール構造を利用することで、プログラム可能なスプライシング制御因子を作り出すことができる。
4.5SH RNAのモジュール構造を利用して、SL1-4.5SHとターゲットとなるエクソンへの相補配列からなるプログラマブル・スプライシング・レギュレーターを作製できるかどうかを検討した。FASのよく知られた代替エクソン(ex)6(FAS-ex6)24とそれに隣接するエクソンを含むミニ遺伝子を調製し、FAS-ex6に相補的な配列に融合したSL1-4.5SHからなるキメラ分子(SL1-4.5SH-asFAS)の活性を試験した(図7Aと7B)。予想通り、SL1-4.5SH-asFASはFAS-ex6のスキッピングを誘導したが、4.5SH RNA単独ではこのミニ遺伝子のスプライシングに影響を与えなかった(図7Aと7B)。キメラ分子の幅広い適用性をさらに確認するために、アンチセンスオリゴヌクレオチドを介したエクソンスキッピング療法の標的エクソンであるMAPT25のエクソン10またはヒトDMDのエクソン53を含むミニ遺伝子も作製した(図7A)26,27。MAPTキメラ(SL1-4.5SH-asMAPT)でも効率的なエクソンスキッピングが誘導されたが(図7C)、DMDキメラを単独で用いた場合は、どれも標的エクソンのスキッピングを誘導できなかった。そこで、DMDキメラのさまざまな組み合わせを試したところ、SL1-4.5SH-asDMD2とSL1-4.5SH-asDMD4を同時に導入したときにスキップ産物が検出されたが、使用したPCRサイクルが高すぎて、スキップしていない産物を定量的に検出することはできなかった(図7E)。SL1-4.5SH-asDMD2が標的とする領域は、デュシェンヌ型筋ジストロフィー(DMD)患者の治療に用いられるアンチセンスモルホリノオリゴヌクレオチド治療薬であるViltolarsen(vltr)の標的と部分的に重なっていることに気づいた28。asDMD2をasDMDvltrに置き換えることで、コンビネーショントランスフェクションのエクソンスキッピング活性がさらに向上し、asDMDvltrとasDMD4からなるハイブリッド標的認識モジュールを持つキメラ(SL1-4.5SH-asDMDvltr4、図7F)を用いたときに最高の効率が得られた。同じ戦略を用いて、マウスDmdのエクソン23のスキップを効率的に誘導するキメラRNA(SL1-4.5SH-asDmd)の作製に成功した(図7Aおよび7F)。
ダウンロード 高解像度画像ダウンロード(1MB)
ダウンロード フルサイズ画像のダウンロード
図7. キメラ4.5SH RNAはデザイン可能なスプライシング制御因子として機能する。
(A)キメラ分子を作るために使われた領域とエクソンスキッピング活性を評価するために使われた戦略を示す模式図。青いバーは、SL1-4.5SHと相補的な標的認識配列からなるキメラ4.5SH RNAが標的とする領域を示す。標的エクソンとそれに隣接するイントロン/エクソン配列を含むミニゲインを、U6プロモーターの制御下でキメラ分子を発現するベクターとともにHEK293細胞に共導入した。
(B、C、F)スプライシングミニゲンと4.5SH-キメラRNAを発現するベクターでトランスフェクトしたHEK293細胞におけるFAS-ex6(B)、MAPT-ex10(C)、Dmd-ex23(F)の代替スプライシング。キメラRNAの発現はノーザンブロットで確認した。FASとDmdについては、SL1-4.5SHをSL1-B1に置き換えると、エクソンスキッピング効率が大幅に低下した。対照的に、MAPTではわずかな減少しか観察されなかった。アスタリスクはバイオアナライザーに現れたヘテロ二重鎖バンドの位置を示す。
(D)スプライシングミニジーンと4.5SH-DMDキメラの異なる組み合わせを発現するベクターをトランスフェクトしたHEK293細胞におけるDMD-ex53の代替スプライシング。この実験で使用したPCR条件は、スキップされていない産物の量を定量的に検出するには大きすぎた。
(E)スプライシングミニジーンと様々な4.5SH-DMDキメラを発現するベクターでトランスフェクトしたHEK293細胞におけるDMD-ex53の代替スプライシングの定量。ハイブリッドキメラSL1-4.5SH-DMDvltr-4は効率的なエクソンスキッピングを誘導した。キメラRNAの発現はノーザンブロットで確認した。
(G)スプライシングミニ遺伝子とSL1-4.5SH-DMDvltr-4および異なる濃度のViltolarsenを発現するベクターでトランスフェクトしたHeLa細胞におけるDMD-ex53の代替スプライシングの定量。キメラRNAのスキップ効率は、最高濃度のViltolarsenのそれと同等であることに注意。SL1-4.5SH-DMDvltr-4の発現はノーザンブロットで確認した。エラーバーは、(B)-(D)および(F)では生物学的3連産物、(G)では生物学的2連産物からの標準偏差を表す。
SL1-4.5SHを介したエフェクター複合体のリクルートが、これらのキメラ分子のエクソンスキッピングに重要であるかどうかを調べるために、SL1-4.5SHをSL1-B1で置換した(SL1-B1-asFAS、SL1-B1-asMAPT、SL1-B1-asDmd、SL1-B1-asDMDvltr4)。FAS、Dmd、DMDについては、置換によってエクソンスキッピング活性の顕著な低下が観察されたが(図7B、7D、7F)、MAPTについては、より緩やかな低下が認められた(図7C)。これらの結果は、これらの合成キメラRNAのエクソンスキッピングが、標的エクソンに応じて多様なメカニズムで作用することを示している。ある種のエクソンでは、キメラRNAとの単純な塩基対形成だけでは不十分で、SL1 4.5SHを通してリクルートされたスプライシング阻害剤の関与が必要である。逆に、塩基対だけで効果的にスキップされるエクソンもあり、これはおそらく、弱い5′および3′スプライス部位と連動して働くエクソニックスプライシングエンハンサーがマスクされるためであろう。
最後に、レポーター系を用いてSL1-4.5SH-asDMDvltr4とViltolarsenのエクソンスキッピング活性を比較した。この実験には、プラスミドとモルフォリノオリゴヌクレオチドの両方で効率的なトランスフェクション効率を示すHeLa細胞を用いた。このハイブリッドキメラ分子のエクソンスキッピング活性は、遺伝子治療に用いられる濃度である10μMのViltolarsenと同様であった(図7G)。
考察
本研究では、4.5SH RNAがasB1の有害なエキソニン化に対する「分子解毒剤」として働き、小型げっ歯類の進化の過程でSINE B1の侵入からトランスクリプトームの完全性を守っていることを証明した。HNRNPCは以前、ポリピリミジン(ポリU)トラクト配列をU2AF2と競合することによって、SINE B1のヒトホモログであるAluエレメントのエクソン化を阻害することが示されている4。HNRNPCの枯渇は、ヒト細胞で外来的に発現する4.5SH RNAのエクソンスキッピング活性に影響を与えなかったことから、げっ歯類と霊長類は、それぞれRNAとタンパク質を介して、SINE B1ファミリーレトロトランスポゾンの有害なアンチセンス挿入からトランスクリプトームを保護する独自のシステムを進化させてきたようである。しかし、BC200転写産物は主に細胞質に存在するため、エクソンスキッピングに関与することはない。多くのSINE由来のノンコーディングRNAが豊富に発現している一方で、核局在の獲得は、小型げっ歯類の系統内で4.5SH RNAがスプライシング制御因子として出現するための重要なステップであった可能性がある。
HNRNPCはアンチセンスAluエレメントの2分割ポリU配列を認識するが、マウスではasB1が1本のポリUストレッチしか持たないため、効率的に働かない可能性がある。我々は、A-ストレッチを欠く3′切断SINE B1のSL1領域に点突然変異を導入することで、非翻訳可能なHnrnpm結合核4.5SH RNAが出現し、これがasB1の有害なエクソン化を打ち消すというモデルを提案する。その後、この有利なノンコーディングRNAを含むゲノム断片が増幅され、大きなタンデム遺伝子クラスターを形成した。このクラスターから転写される豊富な4.5SH RNAは、有毒なasB1を含むエクソンの包含を効率的にブロックし、マウスやラットなどの小型げっ歯類では必須となった。4.5SH RNAの標的認識モジュールがミスマッチ変異を許容できることを考慮すると、4.5SH RNA依存的なasB1含有エクソンのスキップは、HNRNPC依存的なAlu含有エクソンのスキップよりも頑健であると思われる。言い換えれば、Aluのアンチセンス挿入は、HNRNPCによる認識から逃れるためにポリ-Uストレッチに変異を持つ場合、エクソン化される可能性が高い。挿入の数で正規化した場合でも、RefSeqで注釈付けされた遺伝子では、ヒトのAlu含有エクソンはマウスのSINE B1含有エクソンよりも豊富である。
4.5SH RNAの標的認識モジュールはプログラム可能であり、SL1-4.5SHとデザインされた標的認識配列からなるキメラ分子は、目的のエクソンのスキップを誘導することができる。修飾アンチセンスオリゴヌクレオチド(ASO)は、エクソンスキッピングを誘導することで機能的に救済できる変異を持つ疾患を治療する有望な治療薬として台頭してきている。ASOを介した治療の限界は、高用量のオリゴヌクレオチドを繰り返し投与することであり、これは患者にとって大きな負担となりうる。しかし、U7キメラ分子を過剰発現させると、内因性プロセスを阻害してU7 snRNAのプロセシングに異常をきたすことが報告されている34。4.5SH RNAは小さな齧歯類特異的ノンコーディングRNAであり、したがって4.5SHキメラRNAの発現が内因性経路を直接阻害しないことを考慮すると、我々の方法はベクターベースの遺伝子治療に代替的なアプローチを提供する可能性がある。
治療用ASOは制御配列と塩基対を形成してエクソンスキッピングを誘導するが、4.5SH RNAは異なる分子機構を介してエクソンの封入を阻害することが2つの観察から示唆された。第一に、SL1-4.5SHをSL1-B1に置換すると、標的配列との相補性が高まるにもかかわらず、エクソンスキッピング活性が著しく低下した。第二に、標的認識モジュールに完全一致変異を導入すると、4.5SH RNAのエクソンスキッピング活性が低下した。従って、4.5SH RNAを介したエクソンスキッピングには単純な塩基対形成だけでは不十分であり、その完全な活性にはさらなるエフェクタータンパク質が必要である。このメカニズムは、トランス作用するスプライシング制御因子をリクルートするシス要素を含む人工的な二機能性アンチセンスオリゴヌクレオチドを彷彿とさせる35,36。我々は、少なくとも3つのエフェクターRNA結合タンパク質、Hnrnpm、Sfpq、Nonoが、SL1-4.5SHと特異的に会合するが、非機能性SL1-B1とは会合しないことを見出した。HnrnpmはSfpq/Nonoと会合し、特定のエクソンのスキップを誘導することが以前に示されている37。さらに、ゲノムワイドな解析から、このタンパク質を枯渇させると、Hnrnpmと会合したエクソンが優先的に組み込まれ、スプライシングのネガティブレギュレーターとして機能することが明らかになっている19,38。最も代表的なHnrnpm結合モチーフGUGGUGG19がSL1-4.5SHの遠位領域に位置していることを考えると、Hnrnpmは4.5SH RNAに結合する一次エフェクタータンパク質であり、その後にSfpqやNonoを含む追加エフェクター複合体をリクルートしてエクソンスキッピングを誘導するのかもしれない。SfpqとNonoの両方がプレmRNAのイントロン領域に結合することを考えると39,40、4.5SH RNA-タンパク質複合体の形成は、暗号化されたasB1を含むエクソンを、SfpqとNonoが濃縮されているイントロンが濃縮された核内コンパートメント41に勧誘するのかもしれない。あるいは、標的エクソン上に大きなリボ核タンパク質複合体が形成されると、立体障害によって3′スプライス部位の認識が物理的に妨害される。正確な分子機構を明らかにするためには、4.5SH RNAの存在下または非存在下でin vitroのプレmRNAプロセッシングを低温電子顕微鏡で観察するのが興味深い。この方法は、スプライソソームの段階的な形成と反応を明らかにするのに成功した42。
Hnrnpmは高度に保存されたRNA結合タンパク質43であり、長鎖ノンコーディングRNA(lncRNA)を含む様々な種類の細胞内RNAと会合する。この仮説と一致して、PLANEと呼ばれるlncRNAがHNRNPMと結合し、NCORのalternative splicingを制御することが最近示された45。PLANEは標的エクソンの下流のイントロン領域に結合するので、エクソン配列を認識して上流の3′スプライス部位の使用を阻害する4.5SH RNAとは異なる分子機構を利用しているのかもしれない。いずれにせよ、alternative splicingの制御はlncRNAの主要な機能の一つであり、トランスクリプトームの複雑さを増大させる制御のもう一つの層を提供する可能性がある。
研究の限界
我々の研究は、4.5SH RNAの生理学的機能と、SINE B1の毒性エキソニン化に対する分子的解毒剤としての役割について新たな知見を提供するものであるが、いくつかの限界もある。我々の研究は主に小型のげっ歯類、特にマウスに焦点を当てており、ヒトAluで実証されたように、その結果は他の種に直接適用できないかもしれない。加えて、エクソンスキッピングにおける4.5SH RNAの役割のメカニズム的理解はまだ不完全である。我々は、Hnrnpm、Sfpq、Nonoとの相互作用に基づく4.5SH RNAの活性モデルを提案しているが、正確な分子メカニズムはまだ完全には解明されていない。今後、低温電子顕微鏡観察などの研究が進めば、より包括的な知見が得られるかもしれない。4.5SH RNAの標的認識モジュールはプログラム可能であることが示されたが、より広範な応用、特に治療への応用には、より多様な生物系や様々な生理的条件下での厳密な検証が必要である。最後に、我々の研究は、4.5SH RNAと同様にスプライシングを制御するかもしれない他のlncRNAの存在の可能性を示唆しているが、直接的な証拠は不足している。包括的なスクリーニングと機能的アッセイが、同様の制御的役割を持つ他のlncRNAを発見するために必要であろう。
STAR★方法
主要リソース表
試薬またはリソースのソース IDENTIFIER
抗体
Nono A キングに対するマウスモノクローナル抗体 A. Foxより寄贈 PMID: 20881053
Sfpq に対するマウスモノクローナル抗体 Abcam #ab11825 ; RRID: AB_298607
Hnrnpm に対するマウスモノクローナル抗体 Santa Cruz #sc -134360; RRID: AB_2264332
Srsf1 に対するマウスモノクローナル抗体 Invitrogen #32 -4600; RRID: AB_2533080
FLAG FUJI FILMに対するマウスモノクローナル抗体 #014 -22383; RRID: AB_10660291
Nanog に対するマウスモノクローナル抗体 Reprocell #14 -5761-80; RRID: AB_2616320
Smchd1 に対するウサギのポリクローナル抗体 T. Sado から頂いた PMID: 30126901
Smg6 に対するウサギポリクローナル抗体 Abcam #ab87539 ; RRID: AB_10674461
hnRNP-C1/C2 に対するマウスモノクローナル抗体 Merck #R5028 ; RRID: AB_261961
抗αチューブリンに対するマウスモノクローナル抗体 Abcam #ab11304 ; RRID: 297909
細菌株およびウイルス株
4.5SH RNAを発現するレンチウイルス 本試験 N/A
生物学的サンプル
C57BL6/N バックグラウンドで維持された 4.5SH KO マウスの胚。
化学物質、ペプチド、組換えタンパク質
メデトミジン ゼノアック 該当なし
ミダゾラム SANDOZ N/A
ブトルファノール 明治 N/A
プロテイナーゼK サーモフィッシャーサイエンティフィック #25530 -015
Hybond®-N+ ハイブリダイゼーション膜 MERCK GERPN203B
パーフェクトハイブプラス MERCK #H7033
DIG easy Hyb MERCK #11603558001
アルカリホスファターゼ標識抗DIG抗体 MERCK #11093274910
CDPスター MERCK #11685627001
カードKSOM弓道N/A
ノックアウト血清サーモフィッシャーサイエンティフィック #10828010
GMEM メルク #G5154
D-MEM/ハムズF-12 富士フイルム 048-29785
リバートラエース 東洋紡 #TRT -101
プロテアーゼ阻害剤カクテル ナカライ #03969 -21
トリトン-X100 ナカライ #9002 -93-1
Tween-20 ナカライ #9005 -64-5
Trozol Thermo Fisher Scientific #15596026
Trizol-LS Thermo Fisher Scientific #10296010
Dynabeads MyOne Streptoavidin T1 サーモフィッシャーサイエンティフィック #65601
RNase阻害剤 TAKARA #2313A
DTTナカライ #14128 -91
抗FLAG M2-アガロースビーズ MERCK #A2220
重要な市販アッセイ
THUNDERBIRD Next SYBR qPCR Mix 東洋紡 #QPX -201
TruSeq Stranded Total RNA Library Prep Gold ILLUMINA #20020598
ReverTra Ace® qPCR RTマスターミックス(gDNAリムーバー付) 東洋紡 #FSQ -301
GoTaq Green マスターミックス PROMEGA #M712B
DNA1000 キット Agilent NA
ECL-prime GEヘルスケア #RPN2232
寄託データ
WTおよび4.5SH KOマウス由来ES細胞のRNA-Seqデータ この研究 PRJDB13752
オリジナル画像 本研究, Mendeley Data 10.17632/265cd268pz.1
実験モデル 細胞株
HEK293 ATCC CRL-1573
nih3t3 atcc crl-1658
WT胚由来ES細胞 本研究 WT胚由来ES細胞
4.5SH KOマウス由来ES細胞 4.5SH KOマウス由来ES細胞
Neuro2A ATCC CCL-131
実験モデル 生物/系統
C57BL6/N バックグラウンドで維持された 4.5SH KO マウス胚 本研究
オリゴヌクレオチド
qRT-PCR用プライマー、ベクター構築用プライマー、RNA-FISH用プローブ、表S2参照。
ソフトウェアおよびアルゴリズム
HISAT2 http://daehwankimlab.github.io/hisat2/ v.2.2.1
Stringtie https://ccb.jhu.edu/software/stringtie/ v2.1.7
rMATS https://rnaseq-mats.sourceforge.net v4.4.1
svm-bpfinder https://github.com/ComputationalRNABiology/svm-bpfinder N/A
MaxEntScan http://hollywood.mit.edu/burgelab/maxent/download/ N/A
MEME https://meme-suite.org/meme/ V5.4.1
RNAfold2 https://anaconda.org/bioconda/viennarna V2.5.1
リソースの有無
連絡先
リソースおよび試薬のリクエストは、リードコンタクトである中川伸一 (nakagawas@pharm.hokudai.ac.jp) までお願いします。
材料の入手可能性
本研究で作成された全ての材料は、リクエストに応じて入手可能である。
データおよびコードの利用可能性
RNA-seqの生データはPRJDB13752で入手可能。生画像データはhttps://doi.org/10.17632/265cd268pz.1。
本論文ではオリジナルのコードは報告していない。
本論文で報告されたデータを再解析するために必要な追加情報は、要請があれば主任連絡先から入手可能である。
実験モデルと研究参加者の詳細
本研究で開発した4.5SH KOマウスモデルを利用した。C57BL/6Nマウスは、広範な戻し交配のための背景系統として使用した。すべての手順は、北海道大学安全管理部の承認(承認番号2015-079)のガイドラインに従って実施した。麻酔にはmedetomidine-midazolam-butorphanol46を10μl/g体重で腹腔内注射した。マウスはC57BL6/Nと広範囲に戻し交配した。メンデル比の評価には、8週齢以上のオスとメスの成体マウスを用いた。解析のために胚を採取したが、性別は決定しなかった。
方法の詳細
ゲル電気泳動およびサザンブロット解析
マウス胚性線維芽細胞(MEF)をトリプシン処理で採取し、PBSで2回洗浄した後、2×106個の細胞を100μlのPBSに懸濁し、48℃で予熱した。この細胞懸濁液を、あらかじめPBSで加温した等量の1.2%低融点アガロースと混合し、100μlの混合液をウェルにロードしてプラグを作製した。各プラグをNDSK緩衝液(0.5M EDTA、1% N-ラウリルサルコシン、1mg/ml Proteinase K(Thermo、#25530-015)、pH 9.5)中で50℃で一晩インキュベートし、さらに新鮮なNDSK緩衝液中で10時間インキュベートした。0.1mMフェニルメタンスルホニルフルオリドを含むTEで洗浄後、各プラグの3分の1を1×Cutsmart緩衝液(NEB)で平衡化し、37℃で一晩制限酵素で処理した。消化されたDNAは、CHEF-Mapper XAシステム(Bio-Rad)を用いて、TAE中の1%アガロースゲル上で2V/cm、パルス時間5分〜40分、14℃で92時間泳動した(角度:106°)48。従来のゲル電気泳動では、5μgのDNAを制限酵素で消化し、TAE中の0.7%アガロースゲル上で分離した。電気泳動後、ゲルを0.25M HClで30分間処理し、1.5M NaClを加えた0.5M NaOHで変性させ、1.5M NaClを加えた0.5M Tris(pH 7.2)で中和し、20×SSCで平衡化した。DNAを標準プロトコールでHybond-N+(Sigma, #RBN203B )にキャピラリーブロッティングし、DIG easy Hyb (Merck, #11603558001 )中のLNAプローブと製造元の指示に従って60℃でハイブリダイズした。2×SSCで洗浄後、0.2×SSCでストリンジェントに洗浄し、ハイブリダイズしたプローブをアルカリホスファターゼ標識抗DIG抗体(Merck, #11093274910 )とCDP-Star(Merck, #11685627001 )で製造元の指示に従って検出した。化学発光シグナルはChemi Doc Touchイメージングシステム(Biorad)を用いて検出した。
ターゲティングベクターおよびgRNA発現ベクターの構築
ターゲティングベクターを作製するために、相同アーム配列(左アーム:chr6:47,648,963-47,650,909、右アーム:chr6:47,778,356-47,780,356)とloxPおよび制限部位を合成し、プラスミドにクローニングした。相同組換え効率を高めるために、相同アームの末端領域に対してgRNA配列を設計した(図S1AおよびS1 B)。gRNA発現プラスミドを作製するために、表S2に示す2組のオリゴヌクレオチドをアニーリングし、既述のようにpX330-B/BのBbsIおよびBsaI部位にサブクローニングした49。
4.5SH KOマウスの作製
6NK750はC57BL/6N(日本クレア)から樹立したフィーダー依存性ES細胞株であり、既述のように増殖させた51。6NK7 ES細胞(6×106個)を0.8mlのリン酸緩衝生理食塩水(PBS)に懸濁し、Gene Pulser XcellTM(Bio-Rad)を用いて、30μgのターゲティングベクター、各13μgのpX330ベクター、および1.5μgのpPGK-puroプラスミドのサーキュラーフォームを共エレクトロポレーションし、3枚の10cmプレートにプレーティングした。24~48時間後、細胞を1.7μg/mlのピューロマイシンで選択し、その後130μg/mlのG418で1週間選択した。8日目にコロニーを摘出し、ストックした。G418耐性のESクローンを、表S2に示したプライマーを用いてPCR法により、4.5SHクラスターとの相同組換え、およびランダムインテグレーションの非存在についてスクリーニングした。キメラマウスは、ES細胞とICRマウス(日本クレア)の8細胞胚を凝集させて作製し、C57BL/6N雌(日本クレア)と交配してF1ヘテロ接合体マウスを得た。生殖細胞系列の伝達はPCR法で検証し、相同組換えはF1動物の尾から調製したゲノムDNAを用いたサザンブロット分析でさらに検証した。PGK-NeOカセットは、体外受精卵にCre-リコンビナーゼをエレクトロポレーションして除去し、これをレシピエントマウスに移植してフロックス動物を得た。4.5SH KOマウスの遺伝子型決定に用いたプライマーを表S2に示す。遺伝子型決定には以下のPCR条件を用いた:94℃、2分間の1サイクル;94℃、30秒間、57℃、30秒間、68℃、1分間の35サイクル;次いで68℃、5分間。
in situハイブリダイゼーション
E6.5胚から組織切片を調製するために、妊娠マウスをHCMF(10 mM HEPES [pH 7.4]、137 mM NaCl、5.4 mM KCl、0.34 mM Na2HPO4、5.6 mMグルコース)中4%パラホルムアルデヒドで灌流し、さらに剥離した剥皮を4℃で一晩固定した。PBSで2回洗浄後、サンプルをPBS中30%スクロースで凍結保護し、Tissue-Tek OCTコンパウンド(サクラ)に包埋した。培養細胞のin situハイブリダイゼーションでは、細胞をPLLでコートした洗浄済みカバースリップ上で培養し、HCMF中4%パラホルムアルデヒドで4℃で一晩固定した。プローブの調製とin situハイブリダイゼーションは、以前に記載された方法で行った52。簡単に述べると、固定した組織切片または細胞をHClとProteinase Kで処理し、4%PFAで固定し、トリエタノールアミン-HCl緩衝液中、無水酢酸でアセチル化した。灌流による固定とプロテイナーゼK処理は、4.5SH RNAシグナルの検出に不可欠であった。
4.5SH KO ES細胞の樹立と細胞培養
ヘテロ接合体の雌と雄を用いた体外受精で受精卵を得た。胚はKSOM54培地で胚盤胞が拡大するまで4日間培養した。胚盤胞をKSR-GMEM培地中、0.1%ゼラチンでコートした48ウェルプレートに個々にプレーティングし、孵化・増殖させた。培養10日後、内細胞塊(ICM)外生子をフィーダー層を有する24ウェルプレートにプレーティングし、次いで膨張させてストックした。Neuro2A、HEK293、HEK293T細胞は、10%ウシ胎児血清とペニシリン/ストレプトマイシンを添加したDMEM/HamF12の1:1混合液で培養した。
免疫組織化学
細胞をPLLコートしたカバースリップ上で培養し、HCMF中4%PFAで室温10分間固定し、100%メタノール中-20℃で5分間透過処理した。非特異的結合は、TBST(0.05%Tween-20を含むTBS)中の4%スキムミルクでブロックし、続いて一次抗体および二次抗体とインキュベートした。使用した抗体は表S2に示した。蛍光およびDIC画像は、CCDカメラ(DP74)を装備した蛍光顕微鏡(オリンパス、BX51)を用いて得た。
4.5SH発現レンチウイルスの構築
10×タンデム4.5SH発現レンチウイルスを作製するために、サブクローニングに必要な制限部位を持つ、4.5SH遺伝子とこの遺伝子の発現に必要なフランキング配列55を含む遺伝子断片を合成した(表S2)。この遺伝子断片をXhoIとBglIIで消化し、pGEX-4TのXhoI/BamHI部位にサブクローニングした。得られたプラスミドベクターをXhoIとBamHIで消化し、そこに2番目の断片をサブクローニングした。この操作を10個のタンデムリピートがプラスミドにサブクローニングされるまで繰り返し、pGEX4T-10×4.5SHを得た。pGEX-4T-10×4.5SHをSwaIとXhoIで消化して10×4.5SHを含むDNA断片を調製し、これをMluI(blunted)とXhoIで消化したCS-CDF-CG-PRE由来のベクター56にサブクローニングし、pLenti-10×-4.5SHを作製した。簡単に言うと、pLenti-10×4.5SHまたはpLenti-CA-EGFPをパッケージングプラスミドと混合し、FuGENE HD Transfection Reagents(Promega)を用いて、製造元の指示に従ってHEK293T細胞に導入した。24時間後、10μMのフォルスコリンを培養液に添加し、さらに48時間培養した。培養上清を回収し、0.45μmのフィルターで濾過し、20,000gで4時間遠心分離した。ペレットを1/100量の10%FBS含有DMEMに再懸濁し、アリコートを-80℃で凍結保存した。感染には、ウイルスを培養液で1×濃度に希釈し、37℃で一晩ES細胞とインキュベートした。
4.5SH発現の定量化のためのRT-PCR
4.5SHのcDNAは、0.5μgのtotal RNAと0.1μgのnuclear RNAから、ReverTra Ace (100U/μL) (東洋紡、#TRT-101)を用いて、表S2に示したプライマーを用いて、製造者の指示に従って合成した。合成したcDNAを4容量の蒸留水で希釈し、その後のqPCRの鋳型として用いた。qPCR 反応は、Bio-Rad CFX Connect 上で THUNDERBIRD Next SYBR qPCR Mix(東洋紡、#QPX-201)を用い、製造元の指示に従って行った。
細胞分画およびRNA精製
核分画および細胞質分画は、以前に記載されたとおりに行った12。簡単に説明すると、細胞を氷冷したHEPES緩衝生理食塩水(HBSS:10 mM HEPES [pH 7.4]、137 mM NaCl、5.4 mM KCl、0.34 mM Na2HPO4、1 mM MgCl2、1 mM CaCl2、5. 6 mMグルコース)に懸濁し、CSKT(10 mM PIPES [pH 6.8]、100 mM NaCl、300 mM Sucrose、1 mM EGTA、1 mM MgCl2、1 mM DTT、0.1% Triton-X100、1×プロテアーゼ阻害剤(#03969-21、ナカライ製))に5分間懸濁した。細胞懸濁液を700 gで5分間遠心し、さらに12,000 gで5分間遠心して上清を回収し、2容量のTrizol-LS(Thermo Fisher Scientific, #10296010 )で溶解した。核ペレットをCSKTで1回洗浄し、700 gで1分間遠心し、Trizol(Thermo Fisher Scientific, #15596026 )で溶解した。全RNAを回収するため、細胞をHBSSで1回洗浄し、Trizolで溶解した。Trizolで溶解したサンプルは、クロロホルムを加える前に50℃で5分間加熱し、既述のように半抽出RNAを可溶化した57。
RNA配列決定とデータ解析
RNA-seqデータはPRJDB13752から入手可能である。RNA-seqライブラリーは、TruSeq Stranded Total RNA Library Prep Gold(Illumina、#20020598)を用いて、製造元の説明書に従って調製し、HiSeq4000(Illumina)を用いて50bpのシングルエンドリードを生成して配列決定した。得られたfastqファイルは、HISAT2(v.2.2.1)を用いてmm10ゲノム上にマッピングした58。マッピングされた配列リードに基づく転写アセンブリーは、Stringtie(v2.1.7)を用いて行った59。得られたStringtie GTFファイルを参照として、BAMファイルをrMATS(v4.4.1)14を用いて解析し、代替スプライシングアイソフォームの変化を調べた。スプライシングイベントの有意な変化は、偽発見率(FDR)が0.001未満と算出された場合と定義した。濃縮されたモチーフを同定するために、KO細胞に優先的に含まれるエクソンの配列情報を含むファスタファイルを、MEMEを介して解析した15。繰り返し配列の誤検出を避けるため、4 kb以上のエクソンはこの解析から除外した。RNA二次構造はRNAfold2 RNA-Fold.18を用いて予測した。
RT-PCRによる代替スプライシング解析
ReverTra Ace® qPCR RT Master Mix with gDNA Remover (東洋紡, #FSQ -301)を用いて、全RNA(100〜500 ng)をメーカーの指示に従って逆転写した。得られたcDNAをGoTaq Green Master Mix (Promega, #M712B )を用いて20〜23サイクル増幅し、Bioanalyzer (DNA1000, Agilent)を用いて解析した。「スキップ%」は、スキップされたアイソフォームの濃度(nmol/l)を全体(スキップされたアイソフォームと含まれるアイソフォーム)で割ることにより算出した。RT-PCRに使用したプライマーはすべて表S2に示した。
ウェスタンブロット分析
細胞または精製タンパク質を1×Laemmli's SDSサンプルバッファーで溶解し、標準的なSDS-PAGEプロトコルを用いて5-20%グラジエントゲル(フナコシ、#270M)で分離した。Trans-Blot Turbo System(BioRad)を用いてタンパク質をPVDF膜に転写し、メーカーの指示に従ってECL-prime(GE Healthcare, #RBN2232 )で検出した。タンパク質の検出に使用した一次抗体と二次抗体を表S2に示す。化学発光シグナルはChemi-Doc touch(Biorad)を用いて検出した。SMCHD1 に対するポリクローナル抗体は、佐渡隆氏からいただいた62。
スプライシングミニ遺伝子の構築
Slc25a40、CD55、マウスDMD、ヒトDMDのいずれかに由来するDNA断片を、Infusion HDクローニングキット(タカラ、#639648)を用いてアデノウイルスMajor-late(AdV)ミニ遺伝子63のSac II部位に挿入した。AdV-Slc25a40 由来の Split minigene は、FLAG-pcDNA3 の BamHI および XhoI 部位に PCR 断片を挿入して構築した。63 FAS minigene は、Infusion HD cloning kit を用いて、エクソン 5-7 を含む PCR 断片を pcDNA3 の BamHI および XhoI 部位に挿入して構築した。MAPTミニ遺伝子は、エクソン9-1125を含む合成DNA断片をInfusion HDクローニングキットを用いてpcDNA3のBamHIおよびXhoI部位に挿入することにより構築した。配列は表S2に示した。293、293T、またはHeLa細胞を、FuGENE HD Transfection Reagents(Promega)を用いて、製造元の指示に従って、スプライシングミニジーンと発現ベクターで共導入した。
ノーザンブロット分析
全RNA(約0.5μg)を8M尿素を含む変性PAGEで分離した。次にRNAをHybond-N+ (Sigma, #RBN203B )に300 mAの定電流で40分間セミドライ転写した。メンブレンを架橋し、PerfectHyb Plus(Merck、#H7033)中でDIG標識DNAまたはRNAプローブと60℃(RNAプローブ)または42℃(DNAプローブ)でハイブリダイズさせた。2×SSC で洗浄後、0.2×SSC でストリンジェントに洗浄し、ハイブリダイズしたプローブをアルカリホスファターゼ標識抗DIG 抗体(Merck, #11093274910 )とCDP-Star(Merck, #11685627001 )で製造元の指示に従って検出した。化学発光シグナルはImageQuant 800 (Cytiva)を用いて検出した。
siRNAノックダウン
HEK293細胞を12ウェルプレートで培養し、Lipofectamine RNAiMAX(Thermo社、#13778)を用いて、製造元の指示に従って各siRNAを50pmolずつトランスフェクトした。トランスフェクション後72時間で全RNAを抽出し、RT-PCR解析に供した。
RNAプルダウン
RNAプルダウン実験に用いたビオチン化RNAプローブを表S2に示す。RNAプローブはIDTで合成し、1mMの濃度で二重鎖バッファー(IDT)に再懸濁した。3μlのプローブを7μlの二重鎖バッファーと混合し、94℃で90秒間変性させ、85℃で2秒間リフォールディングを60サイクル行った。30μlのDynabeads MyOne Streptoavidin T1(Invitrogen, #65601 )をPBST(PBS、0.1% Triton X100)で2回洗浄し、20μlのPBSTに再懸濁し、リフォールディングしたプローブと4℃で2時間インキュベートした。プローブ結合ビーズをPBSTで2回洗浄し、核溶解液とともに4℃で3時間インキュベートした。核溶解液を調製するために、10cmの培養皿で培養したNeuro2A細胞(約1×108個)を4℃でHBSSで2回洗浄し、1mlの氷冷CSKTで5分間処理した。細胞核をスクレーパーで集め、1.5mlの低タンパクチューブ(Sigma-Aldrich、#Z666548-250EA)に移し、700gで1分間遠心し、CSKTで1回洗浄した。核ペレットを1mlの結合バッファー(PBST、1mM DTT、1×プロテアーゼインヒビター)に再懸濁し、ハンディソニック(タカラトミー、#UR-21P)を用いて4℃で10秒間隔で5回、5秒間超音波処理した。ライセートを12,000 gで10分間遠心し、上清を新しいチューブに移した。ライセートを、30μlのDynabeads MyOne Streptoavidin T1と100μlのStreptoavidin-conjugated Sepharose(Merck, #17 -5113-01)を用いて、4℃で2時間プレクリアした。5μlのRNase阻害剤(Takara, #2313A )を添加した後、清澄化したライセートをプローブ結合ビーズと混合した。ビーズ上の精製タンパク質は、ウェスタンブロッティングの前に、プロテアーゼ阻害剤と1mM DTTを加えたPBSTで1回、PBSTと1mM DTTで2回洗浄した。いくつかの実験では、ビーズを1M NaClを含むPBSTで洗浄した。LC-MS分析用のサンプルを調製するために、ビーズはさらに1mM DTTを含むPBSで2回洗浄した。
CLIP分析
HEK293細胞を氷冷したPBSで2回洗浄し、直ちに254 nmの紫外線(フナコシ、#FS-1500)を120 mJ/cm2、照射距離約5 cmで照射した。細胞を200μlのSDS緩衝液(50mM Tris-HCl [pH 8.0]、1mM EDTA、150mM NaCl、1mM DTT、1% SDS、1% Triton X-100)に懸濁し、20%の振幅で5秒間超音波処理(QSONICA、#Q125)を3回行った。 0]、1 mM EDTA、150 mM NaCl、1 mM DTT、1% Triton X-100、1×プロテアーゼ阻害剤カクテル(Merck、#P8340)およびRNアーゼ阻害剤(Takara、#2313)添加)。その後、細胞を4℃で20分間、13,500rpmで遠心した。免疫沈降のために、15μlの抗FLAG M2-アガロースビーズ(Merck, #A2220 )を抽出液に加え、4℃で2時間、連続的に回転させながらインキュベートした。ビーズを洗浄バッファー(20 mM Tris-HCl [pH 8.0] 1 mM EDTA、150 mM NaCl、1 mM DTT、0.1% SDS、1% Triton X-100)で6回洗浄した。ビーズをHomomix(50 mM Tris-HCl [pH 7.4]、5 mM EDTA、1.5% SDS、300 mM NaCl、1.5 mg/ml proteinase K)に50℃で60分間懸濁し、その後のノーザンブロット分析に供した。
LC-MS分析
アフィニティー精製したタンパク質を1mMジチオスレイトールで90分間還元し、5.5mMヨードアセトアミドで30分間アルキル化し、トリプシンで37℃で一晩消化した。断片化したペプチドをZipTip C18 (Millipore) を用いて脱塩し、真空濃縮機で遠心した。ショットガンプロテオーム解析は、Ultimate 3000 RSLCnanoポンプ(Thermo Fisher Scientific)に接続したFAIMS Proインターフェース(Thermo Fisher Scientific)付きOrbitrap Eclipse Tribrid質量分析計で行った。ペプチドサンプルは、2-24%移動相(アセトニトリル中0.1%ギ酸)のリニアグラジエントを用い、300nl/minで分離した。フルスキャンMSスペクトルは、自動ゲインコントロール(AGC)目標値4×105、分解能120,000で取得した。その後のMS/MSスキャンは、高エネルギー衝突誘起解離(HCD)フラグメンテーションを用いたイオントラップ・モードで、AGCターゲット値1×104、最大注入時間10msで、35%の規格化衝突エネルギーで行った。タンパク質の同定は、Proteome Discoverer Software (version 2.5) (Thermo Fisher Scientific)のSequest HTアルゴリズムを用いて、UniProtマウス参照プロテオームデータベース(UP000000589)を検索することにより行った。システインのカルバミドメチル化は固定修飾とし、メチオニンの酸化とタンパク質のN末端アセチル化は可変修飾とした。データベース検索では最大2つのミス切断を許容し、質量許容範囲はペプチド質量で10 ppm、MS/MSピークで0.6 Daにそれぞれ設定した。ペプチド同定の過程では、偽陽性率<1%を満たすフィルターを適用した。
MS2CPタグRBP発現ベクターの構築
MS2CP融合タンパク質発現ベクター(pLVSIN-CMV_Pur_FLAG-MS2CP)を構築するために、pcDNA5/FRT/TO/i/FLAG/MCP49の3xFLAG-MS2CP-(GGGGS)3リンカー断片をpLVSIN-CMV_Purベクター(タカラ)のEcoRI部位に挿入した。FLAG-MS2CP-NONO、FLAG-MS2CP-SFPQおよびFLAG-MS2CP-HNRNPM発現ベクターを作製するために、ヒトNONOおよびSFPQオープンリーディングフレームはpLVSIN-CMV_Pur_FLAG-MS2CPのXhoI部位を用いて挿入し、ヒトHNRNPMオープンリーディングフレームはpLVSIN-CMV_Pur_FLAG-MS2CPのXbaI部位を用いて挿入した。
Viltolarsenおよびスプライシングレポーターミニ遺伝子のトランスフェクション
スプライシングレポーターミニジーンとモルフォリノオリゴヌクレオチドは同時に導入できないため、順次トランスフェクションした。まず、HeLa細胞にFuGENE HD Transfection Reagents(Promega社製)を用いて、製造者の指示に従ってスプライシングレポーターミニジーンを混合してトランスフェクトした。24時間後、細胞を回収し、Nucleofectorを用いてViltolarsenでエレクトロポレーションした。5.0×105 個の HeLa 細胞を、1μM、2μM、または 10μM の Viltolarsen を含む 100μl Opti-MEM に懸濁し、NucleofectorⅡを用い、プログラム I-013 でエレクトロポレーションを行った。
定量および統計解析
図中のエラーバーは、生物学的複製から得られた標準偏差を表す。各実験の具体的な反復数(二重反復または三重反復)は、対応する図の説明文に詳述している。統計的評価には、Welchのt検定を行い、必要に応じて多重比較を補正するためにBonferroni補正を適用した。
謝辞
抗SMCHD1抗体を提供してくださった佐渡隆博士、抗Nono抗体を提供してくださったArcha Fox博士、原稿を丁寧に読んでくださったAkila Mayeda博士とRyo Inoue博士、英文校正をしてくださったEditage (www.editage.com)、RNA-seqを提供してくださったUC BerkeleyのQB3 Genomicsに感謝する。本研究は、日本学術振興会 科学研究費補助金(基盤研究(A))第16H06276号(AdAMS)、21H05274号および21K19246号(S.N.)、22K05565号(R.Y.)、21H00253号および22H02545号(T.Y.)、20H05784号(S.I.)、23ym0126801号(S.N.)の助成を受けた。
著者貢献
S.N.とR.Y.は実験のデザインと実施、M.K.は4.5SHとES培養物の定量に貢献、中山雄一とI.Y.はES細胞とマウスを用いた実験、I.N.は欠失変異体とViltolarsenを用いた実験を行った。K.A.は4.5SH変異動物の作製とES細胞の樹立を行い、中川陽一はDmdキメラRNAの最適化を行い、S.T.はウェスタンブロット解析とノーザンブロット解析に貢献し、Y.S.とT.K.はパルスフィールドゲル電気泳動実験に貢献し、H.K.-H.とM.O. H.K.-H.およびM.O.は質量分析に、M.M.およびS.I.はRNA-seqライブラリーの調製に、T.Y.およびT.H.はMS2CPタグ付きRBPの調製に貢献した。スプライシングミニゲンとキメラ分子以外の4.5SH KOマウスおよびその他の資料についてはS.N.宛に、R.Y.宛にはR.Y.宛にお送りください。
利益申告
本成果に基づき、R.Y.およびS.N.が特許出願(JP21663/2022-076674およびPCT/JP2023/015828)を行った。
インクルージョンと多様性
私たちは、包括的で多様性のある衡平な研究実施を支持します。
補足情報
すべての補足ファイルをダウンロードする
これは何ですか?
ダウンロード アクロバットPDFファイルのダウンロード (1022KB)
資料S1. 図S1-S6
ダウンロード スプレッドシートのダウンロード(34KB)
表S1. 図2に関連する、4.5SH KO ES細胞で濃縮されたエクソンのrMATS出力とアノテーション。
ダウンロード スプレッドシートのダウンロード (19KB)
表S2. 本研究で使用したプライマーと抗体。
ダウンロード Acrobat PDFファイルのダウンロード (6MB)
資料S2. 論文と補足情報。
参考文献
1
D.A.クラメロフ、N.S.ヴァセツキー
SINEs
Wiley Interdiscip. Rev. RNA, 2 (2011), pp.
CrossRefScopusで表示Google Scholarで表示
2
R. ソレック
新しいエクソンの誕生:そのメカニズムと進化的帰結
RNA, 13 (2007), 1603-1608ページ
CrossRefScopusで見るGoogle Scholarで見る
3
N. セラ、B.メルシュ、N.ガル-マーク、G.レフ-マオール、A.ホッツ-ワーゲンブラット、G.アスト
ヒトとマウスのゲノムにおける転置エレメント挿入の比較解析から、ヒトのトランスクリプトーム形成におけるAluのユニークな役割が明らかになった。
ゲノム生物学, 8 (2007), p. R127
CrossRefScopusで見るGoogle Scholarで見る
4
K. Zarnack, J. König, M. Tajnik, I. Martincorena, S. Eustermann, I. Stévant, A. Reyes, S. Anders, N.M. Luscombe, J. Ule
hnRNP CとU2AF65の直接競合が、Aluエレメントのエクソナリゼーションからトランスクリプトームを守る
Cell, 152 (2013), 453-466ページ
PDFで記事を見るScopusで記事を見るGoogle Scholarで記事を見る
5
J.S.マティック
ロングノンコーディングRNA生物学の現状
ノンコーディング RNA, 4 (2018)
グーグル スカラー
6
J.L.リン、H.Y.チャン
ロングノンコーディングRNA:分子様式から生物機能まで
Annu. Rev.生化学, 89 (2020), pp.283-308
CrossRefScopusで見るGoogle Scholarで見る
7
F. 原田、加藤直樹
マウスおよびハムスター細胞のポリ(A)含有RNAに関連する4.5S RNAの塩基配列
核酸研究, 8 (1980), 1273-1285ページ
CrossRefScopusで見るGoogle Scholarで見る
8
F. 原田, 加藤直樹, 星野英紀
げっ歯類細胞のポリ(A)含有RNAに関連する一連の4.5S RNA
核酸研究, 7 (1979), 909-917ページ
CrossRefScopusで見るGoogle Scholarで見る
9
W. イェリネク, L. ラインワンド
培養チャイニーズハムスター卵巣細胞の核および細胞質ポリ(A)-末端RNAに水素結合した低分子量RNA
細胞, 15 (1978), 205-214ページ
PDFを見る記事を見るScopusで見るGoogle Scholarで見る
10
I.K.ゴゴレフスカヤ、A.P.コバル、D.A.クラメロフ
4.5SH RNAの進化の歴史
Mol. Biol. Evol., 22 (2005), 1546-1554ページ
CrossRefScopusで見るGoogle Scholarで見る
11
L.O.シェーニガー、W.R.イェリネク
4.5S RNAは数百のタンデムに連結した遺伝子によってコードされ、半減期が短く、in vivoではマウス培養細胞の細胞質でポリ(A)末端RNAと水素結合している。
Mol. Cell. Biol., 6 (1986), 1508-1519ページ
スコープで見るGoogle Scholar
12
K. 石田紘一、宮内恭一、木村祐一、水戸正明、岡田茂樹、鈴木忠志、中川聡明
レトロトランスポゾンの挿入とノンコーディングRNA 4.5S RNAHを介した遺伝子発現制御
遺伝子の細胞, 20 (2015), pp.
CrossRefScopusで見るGoogle Scholarで見る
13
D.L.スペクター、A.I.ラモンド
核の斑点
Cold Spring Harb. Perspect. Biol.
Google Scholar
14
S. Shen、J.W. Park、Z.X. Lu、L. Lin、M.D. Henry、Y.N. Wu、Q. Zhou、Y. Xing
rMATS:複製RNA-Seqデータからの頑健かつ柔軟な代替スプライシング差の検出
Proc. Natl. Acad. Sci. USA, 111 (2014), pp.
スコープで見るGoogle Scholar
15
T.L.ベイリー、J.ジョンソン、C.E.グラント、W.S.ノーブル
MEMEスイート
Nucleic Acids Res., 43 (2015), pp.
CrossRefScopusGoogleスカラーで見る
16
T. 黒崎、M.W.ポップ、L.E.マカット
ナンセンスを介したmRNA崩壊による遺伝子発現の質と量の制御
Nat. Rev. Mol. Cell Biol., 20 (2019), 406-420頁
CrossRefスコープで見るGoogle Scholar
17
M.L.ザップ、S.M.ベルゲット
5′スプライス部位の認識に関与する核内因子の証拠
核酸研究, 17 (1989), 2655-2674頁
CrossRefView in ScopusGoogle Scholar
18
R. Lorenz, S.H. Bernhart, C. Höner Zu Siederdissen, H. Tafer, C. Flamm, P.F. Stadler, I.L. Hofacker
ViennaRNAパッケージ2.0
Algorithms Mol. Biol., 6 (2011), p. 26
スコープで見るGoogle Scholar
19
S.C. Huelga, A.Q. Vu, J.D. Arnold, T.Y. Liang, P.P. Liu, B.Y. Yan, J.P. Donohue, L. Shiue, S. Hoon, S. Brenner, et al.
ゲノムワイドな統合解析により、hnRNPタンパク質によるalternative splicingの協調的制御が明らかになった。
Cell Rep., 1 (2012), 167-178ページ
PDFで記事を見るScopusで記事を見るGoogle Scholar
20
A.H.フォックス、中川聡、廣瀬智紀、C.S.ボンド
パラスペックル:長鎖ノンコーディングRNAと相分離が出会う場所
Trends Biochem. Sci., 43 (2018), pp.
PDFで記事を見るScopusで記事を見るGoogle Scholar
21
L.L.チェン、G.G.カーマイケル
ヒト胚性幹細胞におけるインバーテッドリピートを含むmRNAの核内保持の変化:核内ノンコーディングRNAの機能的役割
Mol. 細胞, 35 (2009), 467-478頁
PDFで記事を見るScopusで記事を見るGoogle Scholarで記事を見る
22
D.M.パッソン、M.リー、O.ラッカム、W.A.スタンリー、A.サドウスカ、A.フィリポフスカ、A.H.フォックス、C.S.ボンド
ヒトNONOとparaspeckle protein component 1のヘテロ二量体の構造と核下体形成におけるその役割の解析
Proc. Natl. Acad. Sci. USA, 109 (2012), pp.
CrossRefScopusで見るGoogle Scholarで見る
23
V.J.バードウェル、M.ウィッケンズ
R17コートタンパク質アフィニティー法によるRNAおよびRNA-タンパク質複合体の精製
核酸研究, 18 (1990), 6587-6594頁
CrossRefScopusで見るGoogle Scholarで見る
24
M.P. Paronetto、I. Bernardis、E. Volpe、E. Bechara、E. Sebestyén、E. Eyras、J. Valcárcel
ユーイング肉腫タンパク質によるFASエクソン定義とアポトーシスの制御
Cell Rep., 7 (2014), 1211-1226頁
PDFで記事を見るScopusで記事を見るGoogle Scholar
25
Q. Yu, J. Guo, J. Zhou
エクソン10の正しいスプライシングには、タウエキソン10と11の間に最小限の長さが必要である。
J. 神経化学, 90 (2004), 164-172頁
スコープで見るGoogle Scholar
26
R. スッド、E.T.ゲラー、G.D.シェレンバーグ
アンチセンスによるエクソンスキッピングはタウ蛋白質の発現を減少させる: タウオ病に対する治療の可能性
Mol. Ther. Nucleic Acids, 3 (2014), p. e180
PDFを見る記事を見るCrossRefScopusで見るGoogle Scholar
27
H. 小牧秀樹、永田崇、齋藤知行、増田聡、竹下英治、佐々木雅人、立森秀樹、中村秀樹、青木陽一、武田修一
デュシェンヌ型筋ジストロフィー患者におけるエクソン53スキップのためのアンチセンスオリゴヌクレオチドNS-065/NCNP-01の全身投与
Sci. Transl. Med., 10 (2018)
グーグル スカラー
28
R.R.ロシュミ、横田貴史
デュシェンヌ型筋ジストロフィー治療薬Viltolarsen
ドラッグ・トゥデイ(Barc), 55 (2019), 627-639頁
クロスリーフScopusで表示Google Scholar
29
H. ティードゲ、W.チェン、J.ブロシウス
ヒトBC200 RNAの一次構造、神経特異的発現、樹状突起の位置
J. 神経科学, 13 (1993), 2382-2390頁
CrossRefScopusで見るGoogle Scholarで見る
30
M. 松尾
アンチセンスオリゴヌクレオチドを用いたエクソンスキッピング療法:デュシェンヌ型筋ジストロフィーから広がる精密医療
気象庁, 4 (2021), pp.232-240
Google Scholar
31
D. リー、F.L.マスタグリア、S.フレッチャー、S.D.ウィルトン
アンチセンスオリゴヌクレオチドを介したエクソンスキッピングによる精密医療
Trends Pharmacol. Sci., 39 (2018), 982-994ページ
PDFで記事を見るScopusで記事を見るGoogle Scholar
32
K. Siva, G. Covello, M.A. Denti
神経遺伝性疾患におけるミスプライシングを修正するエクソンスキッピングアンチセンスオリゴヌクレオチド
Nucleic Acid Ther., 24 (2014), 69-86ページ
CrossRefScopusで見るGoogle Scholarで見る
33
A. Goyenvalle, A. Vulin, F. Fougerousse, F. Leturcq, J.C. Kaplan, L. Garcia, O. Danos
U7 snRNAを介したエクソンスキッピングによるジストロフィー筋の救済
サイエンス, 306 (2004), 1796-1799頁
CrossRefScopusで見るGoogle Scholarで見る
34
A. Eckenfelder, J. Tordo, A. Babbs, K.E. Davies, A. Goyenvalle, O. Danos
細胞内プロセシング能力により、アンチセンス導入に利用可能なキメラU7 snRNAの量が制限される
Mol. Ther. Nucleic Acids, 1 (2012), p. e31
PDFを見るCrossRefを見るGoogle Scholarを見る
35
L.A.スコルディス、M.G.ダンクリー、B.ユエ、I.C.エペロン、F.ムントーニ
二機能性アンチセンスオリゴヌクレオチドは、患者線維芽細胞におけるSMN2遺伝子発現を刺激するトランス作用性スプライシングエンハンサーを提供する。
Proc. Natl. Acad. Sci. USA, 100 (2003), pp.
スコープで見るGoogle Scholar
36
J.P. Brosseau, J.F. Lucier, A.A. Lamarche, L. Shkreta, D. Gendron, E. Lapointe, P. Thibault, E. Paquet, J.P. Perreault, S. Abou Elela, et al.
二機能性オリゴヌクレオチドを用いたスプライシングの方向転換
Nucleic Acids Res., 42 (2014), p. e40
CrossRefスコープで見るGoogle Scholar
37
M. マルコ、M. ライヒター、M. パトリヌー-ゲオルゴーラ、A. ギアリス
hnRNP MはPSFおよびp54(nrb)と相互作用し、定義された核構造内に共局在する
Exp. 細胞研究, 316 (2010), 390-400頁
PDFで記事を見るScopusで記事を見るGoogle Scholar
38
E.L. Van Nostrand, P. Freese, G.A. Pratt, X. Wang, X. Wei, R. Xiao, S.M. Blue, J.Y. Chen, N.A.L. Cody, D. Dominguez, et al.
ヒトRNA結合タンパク質の大規模結合・機能マップ
Nature, 583 (2020), 711-719ページ
CrossRefView in ScopusGoogle Scholar
39
S. Zhang, J.A. Cooper, Y.S. Chong, A. Naveed, C. Mayoh, N. Jayatilleke, T. Liu, S. Amos, S. Kobelke, A.C. Marshall, et al.
NONOは神経芽腫におけるスーパーエンハンサー関連GATA2およびHAND2遺伝子のmRNAプロセシングを促進する
EMBO Rep., 24 (2023), Article e54977
スコープで見るGoogle Scholar
40
K. 飯田耕一、萩原雅子、竹内昭夫
多角的バイオインフォマティクス解析により、RNA結合タンパク質SFPQの機能指向的な標的特異性と認識が明らかになった
iScience, 23 (2020), p. 101325
PDFで記事を見るScopusで記事を見るGoogle Scholar
41
S.E.リャオ、O.レゲブ
相分離核スペックル界面におけるスプライシング:モデル
核酸研究, 49 (2021), pp.
CrossRefスコープで見るGoogle Scholar
42
S.M.フィーカ, 永井一郎
スプライソソームの低温電子顕微鏡スナップショット:動的リボ核タンパク質機械の構造的洞察
Nat. Struct. Mol. Biol., 24 (2017), 791-799頁
CrossRefスコープで見るGoogle Scholar
43
K.V.ダタール、G.ドレイファス、M.S.スワンソン
ヒトhnRNP Mタンパク質:リボ核タンパク質におけるメチオニン/アルギニンリッチリピートモチーフの同定
核酸研究, 21 (1993), 439-446ページ
CrossRefScopusで見るGoogle Scholarで見る
44
T. 福永拓也, 岩切淳一, 小野裕之, 濱田雅史
LncRRIsearch: 組織特異的発現および細胞内局在データを統合したlncRNA-RNA相互作用予測のためのウェブサーバー
Front. Genet., 10 (2019), p. 462
スコープで見るGoogle Scholar
45
L. Teng, Y.C. Feng, S.T. Guo, P.L. Wang, T.F. Qi, Y.M. Yue, S.X. Wang, S.N. Zhang, C.X. Tang, T. La, et al.
汎癌lncRNA PLANEはalternative splicing programを制御し、癌病態を促進する
Nat. Commun., 12 (2021), p. 3734
スコープで見るGoogle Scholar
46
S. 河合、高木由美子、金子修一、黒澤崇史
マウスにおけるケタミンに代わる3種類の混合麻酔薬の効果
Exp. Anim., 60 (2011), pp.
CrossRefScopusで見るGoogle Scholarで見る
47
T. 小林稔侍, 野村宗弘, 堀内孝之
Saccharomyces cerevisiaeにおけるリボソームDNAリピートの伸長に必須なDNAシスエレメントの同定
Mol. Cell. Biol., 21 (2001), pp.
スコープで見るGoogle Scholar
48
D.M.スタルツ、M.W.キレン、H.H.ピアース、A.J.ピアース
ヒトリボソームRNA遺伝子クラスターのゲノム構造と遺伝性
ゲノム研究, 18 (2008), pp.
CrossRefScopusで見るGoogle Scholarで見る
49
T. 山崎哲也、S. Souquere、中條忠昭、S. Kobelke、Y.S. Chong、A.H. Fox、C.S. Bond、中川聡、G. Pierron、廣瀬智紀
NEAT1アーキテクチャーlncRNAの機能ドメインは、相分離を介してパラスペックルの集合を誘導する
Mol. Cell, 70 (2018), pp.
PDFで記事を見るScopusで記事を見るGoogle Scholar
50
I. 本田, 荒木和彦, 本田宗一郎, 添田文雄, 申睦夫, 三隅修一, 山村恭一, 高濱和彦
ドーパミントランスポーターを発現する神経細胞のGIRKチャネルを含むGIRK2サブユニットの欠失は、マウスの強制水泳における無動時間を減少させる。
Neurosci. Lett., 665 (2018), pp.
PDFで記事を見るScopusで記事を見るGoogle Scholar
51
M. 中原, 立山秀樹, 荒木雅人, 中潟直樹, 山村和也, 荒木和也
MSM/Msマウス由来Mol/MSM-1胚性幹細胞を用いた遺伝子トラップ突然変異誘発法
Mamm. ゲノム, 24 (2013), pp.
CrossRefScopusで表示Google Scholarで表示
52
M. 水戸, 川口哲也, 廣瀬智紀, 中川聡明
超解像顕微鏡を用いたRNAとタンパク質の同時多色検出
Methods, 98 (2016), pp.
PDFで記事を見るScopusで記事を見るGoogle Scholar
53
K. 荒木健一郎、武田直樹、吉木明宏、小幡洋一郎、中潟直樹、白石哲也、森脇健一郎、山村恭一郎
MSM/Ms株からの生殖細胞系列コンピテント胚性幹細胞株の樹立
Mamm. ゲノム, 20 (2009), pp.
CrossRefView in ScopusGoogle Scholar
54
J.A.ローイッツ、J.D.ビガーズ
着床前胚の培養
Methods Enzymol.
PDFを見る記事を見るScopusGoogle Scholarで見る
55
A.P.コバル、D.A.クラメロフ
5′-フランキング配列は、RNAポリメラーゼIIIによる4.5SH RNA遺伝子の転写に劇的な影響を与えうる。
遺伝子, 446 (2009), 75-80頁
PDFで記事を見るScopusで記事を見るGoogle Scholar
56
H. 三好秀樹、U. Blömer、高橋正樹、F.H. Gage、I.M. Verma
自己不活性化レンチウイルスベクターの開発
J. Virol., 72 (1998), pp.
CrossRefGoogle Scholar
57
T. 中條卓也, 山崎哲也, 川口哲也, 黒坂誠一郎, 匠哲也, 中川聡, 廣瀬隆一郎
核小体関連構築型ノンコーディングRNAの特徴としての異常な半抽出性
EMBO J., 36 (2017), pp.
CrossRefView in ScopusGoogle Scholar
58
D. キム、J.M.パッジ、C.パーク、C.ベネット、S.L.サルツバーグ
HISAT2とHISAT-genotypeを用いたグラフベースのゲノムアライメントとジェノタイピング
Nat. Biotechnol., 37 (2019), pp.
クロスリーフScopusで表示Google Scholar
59
M. Pertea、D. Kim、G.M. Pertea、J.T. Leek、S.L. Salzberg
HISAT、StringTie、Ballgownを用いたRNA-seq実験の転写レベル発現解析
Nat. Protoc., 11 (2016), 1650-1667頁
CrossRefスコープで見るGoogle Scholar
60
J. Shamani、S.H. Kazakoff、I.M. Armean、W. McLaren、M.T. Parsons、B.A. Thompson、T.A. O'Mara、S.E. Hunt、N. Waddell、A.B. Spurdle
MaxEntScanを使用してバリアントのスプライス原性を予測するEnsembl Variant Effect Predictorのプラグイン
バイオインフォマティクス, 35 (2019), pp.2315-2317
クロスリーフScopusで表示Google Scholar
61
A. コルベロ、M.ハレッガー、C.W.スミス、E.アイラス
分岐点特性とalternative splicingのゲノムワイドな関連性
PLOS Comput. Biol., 6 (2010), Article e1001016
CrossRefScopusで見るGoogle Scholarで見る
62
Y. 榊原由紀夫、長尾和久、M. Blewitt、佐々木秀樹、小布施忠男、佐渡多喜雄
マウスにおけるX不活性状態の維持に必要なエピジェネティック状態の確立におけるSmcHD1の役割
発生, 145 (2018)
Google Scholar
63
R. 吉本、片岡直樹、大川和彦、大野正人
スプライシング後のラリアート-イントロン複合体の単離とキャラクタリゼーション
核酸研究, 37 (2009), pp.
CrossRefView in ScopusGoogle Scholar
引用 (0)
11
リードコンタクト
要旨を見る
© 2023 Elsevier Inc.
エルゼビアロゴとワードマーク
ScienceDirectについて
リモートアクセス
ショッピングカート
広告掲載
お問い合わせとサポート
利用規約
プライバシーポリシー
当サイトではクッキーを使用しています。クッキー設定
このサイトのすべてのコンテンツ: Copyright © 2023 Elsevier B.V., its licensors, and contributors. テキストマイニング、データマイニング、AIトレーニング、および同様の技術に関するものも含め、すべての権利はエルゼビアに帰属します。すべてのオープンアクセスコンテンツには、クリエイティブ・コモンズのライセンス条項が適用されます。
RELXグループホームページ
フィードバック
この記事が気に入ったらサポートをしてみませんか?