
初心者の菌叢解析 Qiime2で解析(6) ファイルの準備編 ~classifierファイル~

初心者の菌叢解析シリーズの6つ目のnoteです。是非全部読んでください。
1.Qiime2を動かすのに必要なファイル
以前の記事でも記載したのですが、必要なファイルが4つあります。
1. Sequenceファイル(.fastqもしくは.fastq.gz)
2. manifestファイル(.csv)
3. sample-metadataファイル(.txt)
4. classifierファイル(.qza)
今回は4つ目のclassifierファイルの準備を行います。
2. classifierファイルとは
classifierファイルは得られたシーケンス情報を元に、そのリードに菌種を当てはめていく為の分類をコントロールするファイルです。
配列と菌種の組み合わせが記されており、日本語では「分類器」と翻訳されると思います。
classifierファイルは菌叢の分類にダイレクトに影響する部分になります。
ですので、自分の菌叢解析にあったclassifierファイルを作成するのが最もよいです。例えばターゲットにしている領域に差がある場合もありますし、使用しているプライマー配列が異なる場合もあります。
自作する場合は以下の投稿を参考にしてください。
3.classifierファイルのダウンロード
classifierファイルのダウンロードは非常に簡単で、Qiime2のホームページからダウンロードが可能です。
まずはQiime2 docsを開きます。

左上の赤枠からバージョンを選びます。同じように進めていれば「Version 2021.2」を開きます。必要に応じて適切なバージョンを選んでください。

最新のバージョンでないと上のようなお知らせが出ますので、右の赤い方をクリックします。
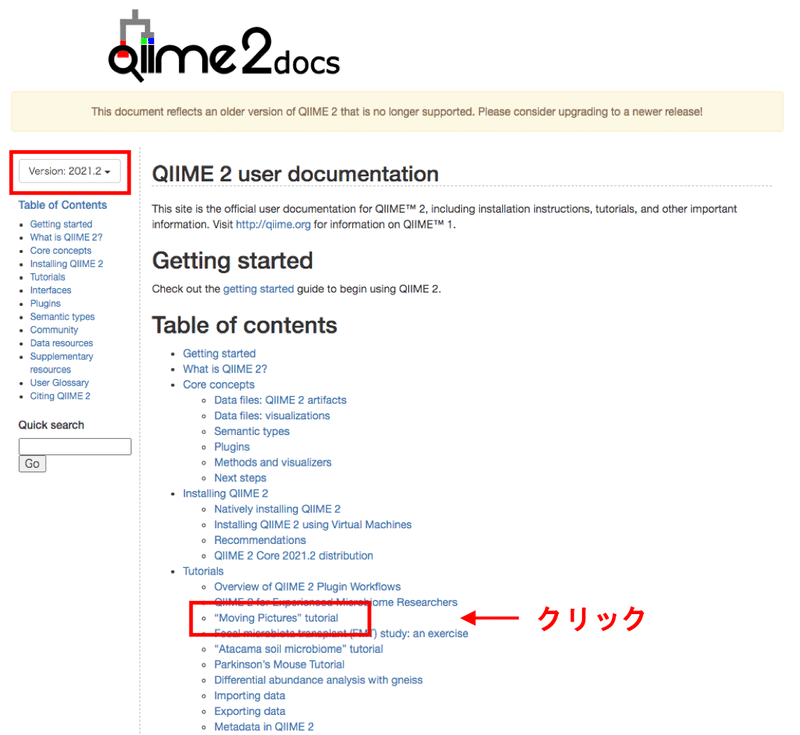
左上のバージョンが目的の物になっていることを確認してください。その後、「”Moving Picture" tutorial」をクリックします。

下の方にスクロールしていくと「Taxonomic analysis」という項目があります。赤枠の「wget」をクリックし、表示されるコマンドをコピーします。
$ wget \
-O "gg-13-8-99-515-806-nb-classifier.qza" \
"https://data.qiime2.org/2021.2/common/gg-13-8-99-515-806-nb-classifier.qza"ターミナルを起動し、フォルダを選択するコマンド「cd」と打ち込みます。「cd」の後にはスペースを入れてください。

その後、目的フォルダ「Qiime2_test」のフォルダをドラッグ&ドロップして、絶対パスの入力を行います。
$ cd /Users/ユーザー名/Desktop/Qiime2_test 私は上記のようなコマンドになりました。
Enterキー(returnキー)を押してフォルダ(ディレクトリ)を移動します。
次にフォルダ(ディレクトリ)内のファイルを確認するコマンド「ls」を入力すると以下の様な返答が得られると思います。
$ ls↓
$ ls
manifest.csv sample-metadata.txt sequence全く同じ内容で進めていれば今まで作成したファイルとフォルダの名前が表示されると思います。ここで、「wget」以下のコマンドを入力します。
$ wget \
-O "gg-13-8-99-515-806-nb-classifier.qza" \
"https://data.qiime2.org/2021.2/common/gg-13-8-99-515-806-nb-classifier.qza"ダウンロードが進み以下の様な表示が出ます。

そうしたら、もう一度「ls」を打ち込みます。
$ ls
gg-13-8-99-515-806-nb-classifier.qza sample-metadata.txt
manifest.csv sequence「gg-13-8-99-515-806-nb-classifier.qza」という名前のclassifierファイルが追加されました。
実際にフォルダを開いてみると、しっかりと保存されています。

これでclassifierファイルの準備が出来ました。
4.ダウンロードしたclassifierファイルについて
「gg-13-8-99-515-806-nb-classifier.qza」はQiime2のチュートリアルで提供されているclassifierファイルになります。ですので、自身の解析におけるclassifierファイルとして不適切な場合は、専用のclassifierファイルを自作する必要があります。
チュートリアル中の説明文を見ると、
「Greengenes 13_8というデータを用いて、99%の配列一致で菌種を同定する様に作成したclassifierファイル」ということになります。
また、「16s rRNAのV4領域を515Fと806Rという2つのユニバーサルプライマーで増幅した250 bpの領域を菌種分類のターゲットとしている」とも記載されています。
つまり、V1-V2領域を狙って菌叢解析を行っている場合は今回のclassifierファイルは使用できません。さらに、V3-V4領域(約500 bp)をターゲットにしている場合も今回のclassiierファイルでは使用は出来るが十分な能力を発揮することが出来ません。
注釈の部分にもありますが、自身の使用したプライマーやPCR産物のサイズ等のシーケンスパラメータを元にclassifierファイルを作成すると、最適な分類が可能ということになります。
ですので、将来的にはclassifierを自作することを強くお勧めします。
再度になりますが、classifierファイルの作成方法については以下の記事で紹介しています。
以下の方の投稿ですと、Qiime2の起動の仕方が若干違いますが、その他は同じですので、「docker」の部分を「conda」に置き換えて進めてください。
次回はQiime2で解析を行います。
ここまで読んでいただきありがとうございました。
この記事が気に入ったらサポートをしてみませんか?