解析パイプライン構築のためのSRAの利用

はじめに
ハイスループットシーケンサーの出力データはFASTQ形式に変換されており、最終的にFASTAファイル形式に変換して取り扱うことが大半かと思います。
FASTAファイルは1行目に” > ”から始まる配列の情報。2行目からは塩基配列のフォーマットになっています。
>KY471393.1_Xenotoca variata
TAAAAGCTTGGTCCTGACTTTTCTATCAGCTCTAGCAAAATTTA . . .
画像情報やシグナル情報が集約されているbclファイルから変換された、配列やクオリティスコアの情報が記載されているFASTQは1行目に"@"の後に配列の名称、2行目に塩基配列、3行目に"+"、4行目にクオリティスコアというフォーマットになっています。
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAA
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>
私はbclファイルからFASTQへの変換はしたことありませんが、サンプルシートなど間違ってしまった場合は、この過程で修正できるみたいですね。(絶対に間違えられないものだと最近まで思っていました)
実験で使用されたこれらのデータは最終的に論文発表時にNCBIやDDBJなどの公共データベースに登録する必要があります。なので、興味のある論文に記載されているaccession IDをもとに登録されているデータベースから入手することによって解析を再現したり、自身の研究に活かしたりすることが出来ます。
今回は自身で作成したパイプラインが使えるのか試したいので、実際に論文で使用されたデータをダウンロードして解析に使えるFASTQ形式に変換することを目的とします。FASTQ形式なら私でも気軽に解析に使うことが出来るので。
実際にやってみる
最近発表された甲殻類のユニバーサルプライマーに関する論文のシーケンスを取得したいと思います。
MiSeqにて解読されたデータでDDBJに登録されています。
・DRA008193 / DRR172676
"DRA008193"はプロジェクトの情報が集約されています。その横はDR"R"で始まっているのでシーケンスデータが入っているFASTQファイルの本体です。シーケンスの日付やリード数、総塩基情報などが記載されています。その他にはDRP, DRX, DRSなど計5種類のフォーマットがあります。
ブラウザでDDBJと検索していただき、国立遺伝学研究所のページに進みます。トップの検索がBI-DDBJ Web SitesになっているのでそれをSequence Readj Archiveに変えて検索窓に欲しいDRR IDを記入して検索します。
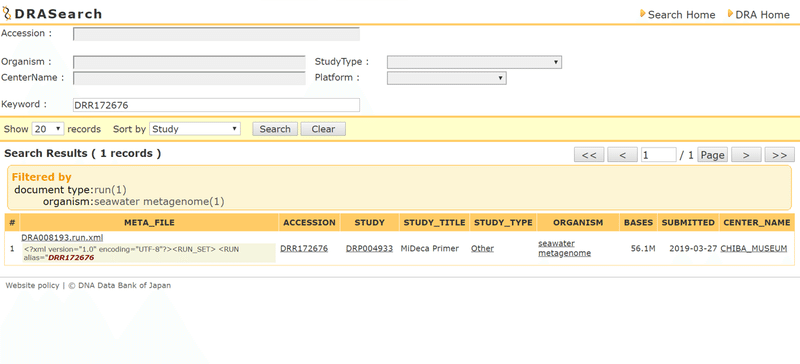
検索したIDに関する情報がでてきました。
Runの結果が欲しいので、DRR172676から次のページに飛びます。

最上段に"FASTQ"とあるのでそこからさらにリンク先へ飛びます。

2つの.fastq.bz2ファイルがありますね。これをダウンロードした後、コマンドラインにてfastqファイルに変換します。
私はWindows環境でUbuntuをインストールして解析を行っています。Windows Subsystem for Linuxを有効にしていると使えます。
bzip2 -d DRR172676_1.fastq.bz2.bzip2形式はbzip2コマンドを使用すると解凍することが出来ます。
これで解析に必要なFASTQファイルが手に入りました。
メタバーコーディング用のデータベースの作成なども試行錯誤しながらやっていますので、またどんな感じでやっているか上げますね。
最後まで読んでいただいてありがとうございます。
Reference pages.
https://bi.biopapyrus.jp/rnaseq/fastq-data/