適応免疫の原理と治療への応用

Principles and therapeutic applications of adaptive immunity
適応免疫の原理と治療への応用
元→Principles and therapeutic applications of adaptive immunity: Cell
Summary
Adaptive immunity provides protection against infectious and malignant diseases.
適応免疫は、感染症や悪性疾患に対する防御を提供します。
These effects are mediated by lymphocytes that sense and respond with targeted precision to perturbations induced by pathogens and tissue damage.
これらの効果は、病原体や組織損傷によって誘発される混乱を感知し、標的とした精度で応答するリンパ球によって媒介されます。
Here, we review key principles underlying adaptive immunity orchestrated by distinct T cell and B cell populations and their extensions to disease therapies.
ここでは、異なる T 細胞と B 細胞集団によって調整される適応免疫の基礎となる重要な原理と、それらの疾患治療への拡張について概説します。
We discuss the intracellular and intercellular processes shaping antigen specificity and recognition in immune activation and lymphocyte functions in mediating effector and memory responses.
免疫活性化における抗原の特異性と認識を形成する細胞内および細胞間のプロセス、およびエフェクターおよび記憶応答を媒介するリンパ球の機能について説明します。
We also describe how lymphocytes balance protective immunity against autoimmunity and immunopathology, including during immune tolerance, response to chronic antigen stimulation, and adaptation to non-lymphoid tissues in coordinating tissue immunity and homeostasis.
また、免疫寛容時、慢性抗原刺激への反応、組織免疫と恒常性の調整における非リンパ組織への適応時など、リンパ球が自己免疫や免疫病理に対する防御免疫のバランスをどのようにとっているかについても説明します。
Finally, we discuss extracellular signals and cell-intrinsic programs underpinning adaptive immunity and conclude by summarizing key advances in vaccination and engineering adaptive immune responses for therapeutic interventions.
最後に、適応免疫を支える細胞外シグナルと細胞固有のプログラムについて議論し、ワクチン接種と治療介入のための適応免疫応答の操作における重要な進歩を要約して結論とします。
A deeper understanding of these principles holds promise for uncovering new means to improve human health.
これらの原則をより深く理解することで、人間の健康を改善するための新しい手段が発見される可能性があります。
Introduction
Adaptive immunity is essential for host protection from infectious and malignant diseases but also contributes to autoimmune and inflammatory disorders under pathophysiological conditions.
適応免疫は、感染症や悪性疾患から宿主を守るために不可欠ですが、病態生理学的条件下では自己免疫疾患や炎症性疾患にも寄与します。
The adaptive immune system is the collection of cells, factors, and effector mechanisms that, through specialized receptors, recognize and respond to specific antigens, which can be derived from entities outside the body (e.g., pathogens and allergens) or within the body itself (e.g., tumors and self-tissues).
適応免疫システムは、特殊な受容体を介して特定の抗原を認識して応答する細胞、因子、エフェクター機構の集合体です。 これは、体外の実体(病原体やアレルゲンなど)または体自体の内部(腫瘍や自己組織など)に由来する可能性があります。
The function of the immune system is analogous to the nervous system, with evolved means of sensing, responding to, and remembering the world.
免疫系の機能は神経系に似ており、世界を感知し、反応し、記憶する手段が進化しています。
The specificity and delayed activation of adaptive immunity contrast with the more rapid and relatively non-specific innate immune response.
適応免疫の特異性と活性化の遅延は、より迅速で比較的非特異的な自然免疫応答とは対照的です。
Further, the adaptive immune system is defined by the emergence of immune memory, the remarkable capacity of lymphocytes to rapidly and precisely respond to a pathogen-derived antigen they encountered before, thereby mediating improved (or complete) protection from re-infection.
さらに、適応免疫系は、免疫記憶の出現によって定義されます。免疫記憶とは、以前に遭遇した病原体由来の抗原に迅速かつ正確に応答するリンパ球の顕著な能力であり、それによって再感染からの防御の向上(または完全な)を媒介します。
To accomplish this, the adaptive immune system utilizes an array of diverse “professional” immune cell types that act as individual but interdependent effectors, along with numerous critical interactions with stromal and parenchymal cells throughout tissues.
これを達成するために、適応免疫系は、組織全体の間質細胞および実質細胞との多数の重要な相互作用とともに、個別ではあるが相互依存するエフェクターとして機能する一連の多様な「専門的な」免疫細胞タイプを利用します。
The core cellular players in adaptive immunity are lymphocytes, specifically T cells and B cells (Figure 1A).
適応免疫における中心的な細胞プレーヤーはリンパ球、特に T 細胞と B 細胞です (図 1A)。
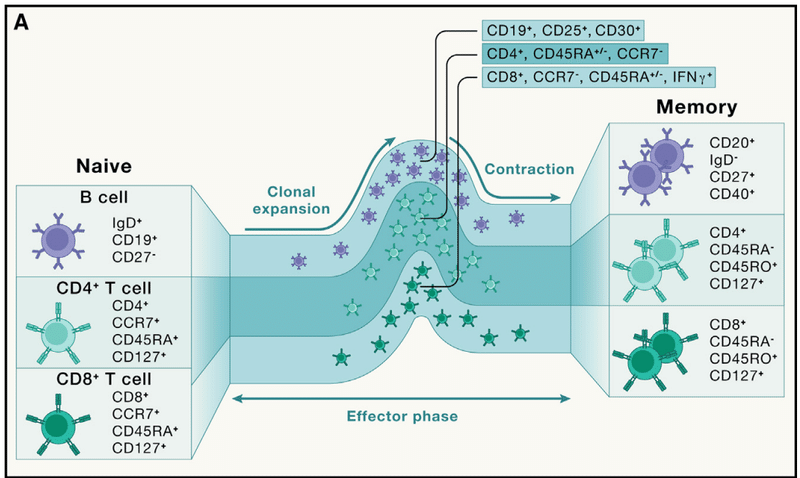
Conventional αβ T cell populations are further classified as CD4+ helper T cells and CD8+ cytotoxic T cells.
従来のαβ T 細胞集団は、さらに CD4+ ヘルパー T 細胞と CD8+ 細胞傷害性 T 細胞に分類されます。
CD4+ T cells exert multiple effector functions, mediated by both soluble factors and cell-cell interactions.
CD4+ T 細胞は、可溶性因子と細胞間相互作用の両方を介して複数のエフェクター機能を発揮します。
CD8+ T cells act primarily through the killing of specific target cells.
CD8+ T 細胞は、主に特定の標的細胞を殺すことによって作用します。
B cells secrete soluble effector molecules called antibodies and can also function as antigen-presenting cells (APCs), which present specific antigens to T cells.
B 細胞は、抗体と呼ばれる可溶性エフェクター分子を分泌し、T 細胞に特定の抗原を提示する抗原提示細胞 (APC) としても機能します。
Antibodies bind target antigens with high affinity and interfere with numerous pathogenic processes, such as blocking the attachment of pathogens to host cells and tissue surfaces, among other functions.
抗体は高い親和性で標的抗原に結合し、機能の中でも特に宿主細胞や組織表面への病原体の付着の阻止など、数多くの病原性プロセスを妨害します。
This process prevents pathogen entry into cells, which can provide sterilizing protection against infection from obligate intracellular pathogens.
このプロセスは病原体の細胞への侵入を防ぎ、絶対的な細胞内病原体からの感染に対する滅菌保護を提供します。
Antibody binding to pathogen surfaces or infected cells also “flags” the cells as targets for innate immune cell-mediated killing, thereby promoting pathogen clearance.
病原体表面または感染細胞に結合する抗体は、自然免疫細胞を介した殺傷の標的として細胞に「フラグ」を立て、それによって病原体の除去を促進します。
Communication between these lymphocytes occurs via two major modalities: secretion of soluble proteins such as cytokines and chemokines and surface ligand-receptor interactions.
これらのリンパ球間のコミュニケーションは、サイトカインやケモカインなどの可溶性タンパク質の分泌と、表面リガンドと受容体の相互作用という 2 つの主要な様式を介して行われます。
These signals alter the functional state of the receiving (and sometimes the sending) cell.
これらの信号は、受信 (場合によっては送信) 細胞の機能状態を変更します。
Collectively, these cell populations, soluble mediators, surface receptors, and their ligands comprise a network of networks, which are overlaid to generate the emergent functions classified as adaptive immunity.
これらの細胞集団、可溶性メディエーター、表面受容体、およびそれらのリガンドは集合的にネットワークのネットワークを構成します。 これらが重ね合わされて、適応免疫として分類される創発機能が生成されます。
Intrinsic redundancy and robustness are key features of the system that maintains function even when deleterious lesions hinder one component.
本質的な冗長性と堅牢性は、有害な損傷によって 1 つのコンポーネントが妨げられた場合でも機能を維持するシステムの重要な特徴です。
Conversely, numerous feedback loops and inhibitory interactions with immune suppressive populations are also wired in, thereby providing an essential balance that restricts self-reactive and potentially damaging responses to preserve homeostasis and prevent immunopathology.
逆に、多数のフィードバック ループや免疫抑制集団との抑制的相互作用も配線されています。 これにより、自己反応性や潜在的に有害な反応を制限し、恒常性を維持し、免疫病理を防ぐ重要なバランスが提供されます。
In this review, we summarize distinct features of adaptive immunity, including how lymphocytes are generated to recognize specific immunological threats (hereafter called threats) and initiate adaptive immunity.
このレビューでは、特定の免疫学的脅威 (以下、脅威と呼びます) を認識し、適応免疫を開始するためにリンパ球がどのように生成されるかなど、適応免疫の明確な特徴を要約します。
We then describe how lymphocytes specialize and adapt to antigen stimulation in both lymphoid and non-lymphoid tissues, including discussing the additional immune stimuli and signaling processes that cooperate with antigen receptor activation to orchestrate adaptive immunity.
次に、リンパ球がリンパ組織と非リンパ組織の両方でどのように特化し、抗原刺激に適応するかを説明します。 これには、抗原受容体の活性化と協力して適応免疫を調整する追加の免疫刺激とシグナル伝達プロセスについての議論も含まれます。
Importantly, throughout these discussions, the regulatory mechanisms that exist to prevent immunopathology and maintain or re-establish homeostasis are described.
重要なのは、これらの議論を通じて、免疫病理を予防し、恒常性を維持または再確立するために存在する制御機構について説明していることです。
Finally, we summarize how adaptive immune responses are harnessed or tailored for therapeutic benefit, including in vaccination against infectious diseases and immune engineering for interventions against cancer and immune-mediated disorders, and close with discussions on emerging and future directions in the field.
最後に、感染症に対するワクチン接種や、がんや免疫介在性疾患に対する介入のための免疫工学など、適応免疫応答がどのように利用され、あるいは治療効果に合わせて調整されているかを要約し、この分野で新たに生まれつつある将来の方向性についての議論で締めくくります。
How do adaptive immune cells “see” the world?
適応免疫細胞は世界をどのように「認識」しているのでしょうか?
B cells and T cells are the essential effectors of adaptive immunity and are exquisitely tuned to target specific pathogens.
B 細胞と T 細胞は適応免疫の必須のエフェクターであり、特定の病原体を標的にするように絶妙に調整されています。
A cascade of events precedes lymphocyte activation and expansion to ensure that their potent effector function is focused on true pathogenic threats.
リンパ球の強力なエフェクター機能が真の病原性脅威に確実に集中するように、一連のイベントがリンパ球の活性化と増殖に先立って行われます。
Here, we discuss the stepwise formation of a primary response and the unique generation of antigen receptors that define the adaptive lymphocyte lineage and mediate antigen recognition.
ここでは、一次応答の段階的な形成と、適応リンパ球系統を定義し抗原認識を媒介する抗原受容体の独自の生成について説明します。
The broad diversity of antigen receptors must be regulated through selection events to limit the development of responses that may damage the host.
宿主に損傷を与える可能性のある応答の発生を制限するには、選択イベントを通じて抗原受容体の広範な多様性を制御する必要があります。
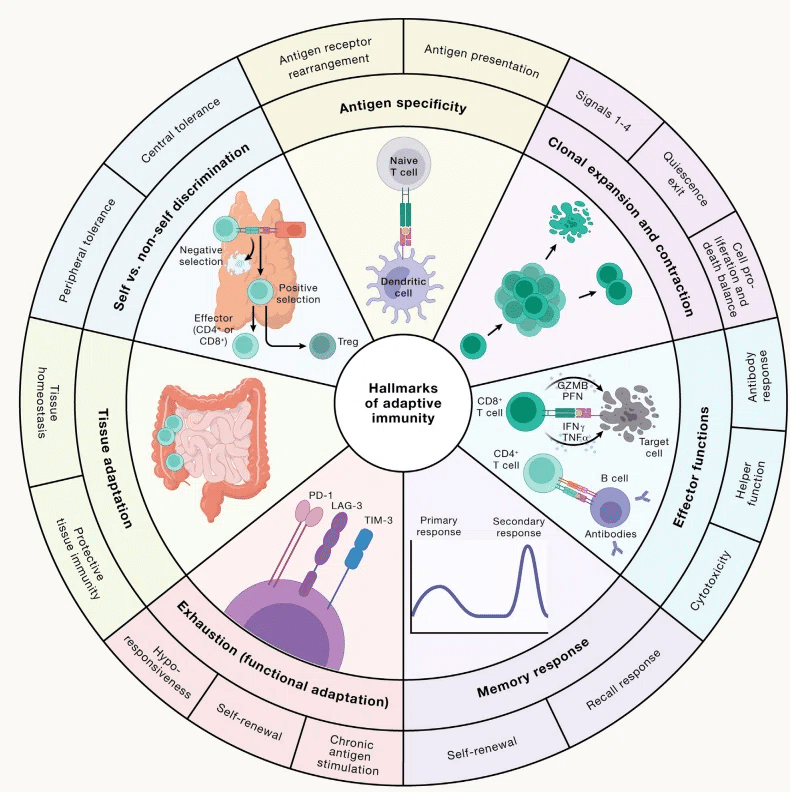
Overview of adaptive immune responses
適応免疫応答の概要
An initial encounter with antigen results in the priming of adaptive immune cells, which subsequently develop into effector cells for immune defense and memory cells to mediate immune memory.
抗原との最初の遭遇により適応免疫細胞がプライミングされ、その後、免疫防御のためのエフェクター細胞と免疫記憶を媒介する記憶細胞へと発達します。
To understand this process, it is helpful to consider the course of a primary immune response (Figure 1B).
このプロセスを理解するには、一次免疫応答の経過を考慮することが役立ちます (図 1B)。
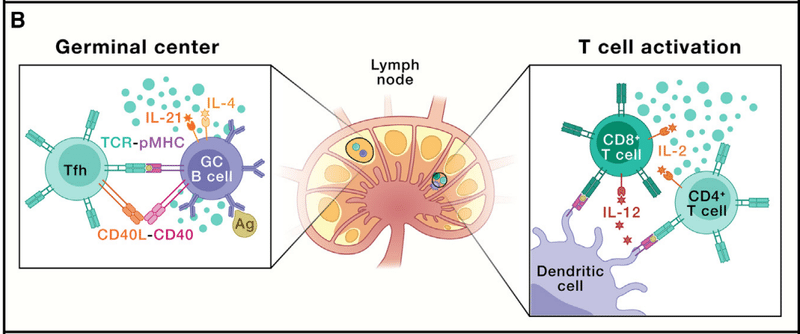
For example, after initial infection by a respiratory virus, epithelial cells and local innate immune cells respond over the course of hours to days to attempt control of the pathogen.
たとえば、呼吸器ウイルスによる最初の感染後、上皮細胞と局所自然免疫細胞は数時間から数日かけて応答し、病原体の制御を試みます。
The lymphocytes that will eventually target this infection are in a quiescent state, circulating through the blood and lymphatics.
やがてこの感染症の標的となるリンパ球は今は静止状態にあり、血液およびリンパ管中を循環しています。
To initiate this response, dendritic cells (DCs, a type of APCs) and the lymphatic vessels traffic viral antigens to local lymph nodes.
この反応を開始するために、樹状細胞 (DC、APC の一種) とリンパ管がウイルス抗原を局所リンパ節に輸送します。
Here, by secreting chemokines, these DCs will attract naive CD4+ and CD8+ T cells.
ここで、これらの DC はケモカインを分泌することにより、ナイーブ CD4+ および CD8+ T 細胞を誘引します。
T cells will then “test” whether they recognize any of the virus-derived antigens “presented” on the DC surface in complex with a host molecule called the major histocompatibility complex (MHC; in humans, these proteins are also called human leukocyte antigens [HLAs]).
次に、T細胞は、主要組織適合性複合体と呼ばれる宿主分子と複合体を形成してDC表面に「提示」されるウイルス由来抗原を認識するかどうかを「テスト」します。 (MHC; ヒトでは、これらのタンパク質はヒト白血球抗原 [HLA] とも呼ばれます)。
T cells “see” the peptide-MHC (pMHC) complex via their antigen receptor, called the T cell receptor (TCR), with co-receptors CD4 and CD8 defining T cell subsets recognizing pMHC antigens via different MHC molecules (class II versus class I, respectively).
T細胞は、T細胞受容体(TCR)と呼ばれる抗原受容体を介してペプチド-MHC(pMHC)複合体を「認識」します。 異なるMHC分子を介してpMHC抗原を認識するT細胞サブセットを定義する共受容体CD4およびCD8を伴います (それぞれクラス II とクラス I)。
Antigen receptors are surface proteins that are the essential sensors of adaptive immune recognition.
抗原受容体は、適応免疫認識の必須センサーである表面タンパク質です。
Within an organism, each T cell, to a first approximation, contains a unique TCR.
生物体内では、各 T 細胞には、多かれ少なかれ、固有の TCR が含まれています。
From any given virus, between 5 and 50 target pMHCs may be recognized, with a corresponding ∼250–25,000 naive T cells capable of recognition and becoming activated by binding with an avidity that triggers sufficient TCR signaling.
任意のウイルスから、5 ~ 50 個の標的 pMHC が認識され、対応する約 250 ~ 25,000 個のナイーブ T 細胞が認識され、十分な TCR シグナル伝達を引き起こす結合力で結合して活性化されます。
If a TCR recognizes pMHC on a DC in the appropriate inflammatory conditions, the T cell becomes activated to initiate the subsequent adaptive immune response.
適切な炎症状態において、TCR が DC 上の pMHC を認識すると、T 細胞が活性化されて、その後の適応免疫応答が開始されます。
For naive T cells, TCR activation leads to an exit from the quiescent state via the activation of downstream signaling pathways and metabolic reprogramming.
ナイーブ T 細胞の場合、TCR の活性化は、下流のシグナル伝達経路の活性化と代謝の再プログラミングを介して静止状態からの脱出につながります。
Massive transcriptional, epigenetic, and translational events promote altered surface protein expression and secretion of cytokines and chemokines, which recruit more naive T cells to the local lymph node to permit additional antigenic “screening.”
大規模な転写、エピジェネティック、翻訳イベントにより、表面タンパク質の発現変化とサイトカインとケモカインの分泌が促進され、より多くのナイーブ T 細胞が局所リンパ節に動員され、追加の抗原「スクリーニング」が可能になります。
Following quiescence exit, a replication program called clonal expansion is initiated.
静止状態の終了に続いて、クローン拡張と呼ばれる複製プログラムが開始されます。
Consequently, there is a pronounced accumulation of antigen-specific T cells that are normally present at low and variable frequencies, thereby heightening the responses against rapidly dividing pathogens.
その結果、通常は低い頻度で存在する抗原特異的 T 細胞が顕著に蓄積し、急速に分裂する病原体に対する反応が高まります。
This replication proceeds via a programmed timer partly regulated by the transcription factor Myc, with the cell undergoing replication as many times as possible within the timer window.
この複製は、転写因子Mycによって部分的に制御されるプログラムされたタイマーを介して進行し、細胞はタイマーウィンドウ内でできるだけ多く複製を受けます。
T cells, once they exit the quiescent state, have among the fastest division times of healthy cells in the mammalian body (estimates of ∼10 h for CD4+ and 6–8 h for CD8+ T cells).
T 細胞は、一旦静止状態から抜け出すと、哺乳動物の体内で健康な細胞の中で最も速い分裂時間になります (CD4+ T 細胞の場合は約 10 時間、CD8+ T 細胞の場合は 6 ~ 8 時間と推定されます)。
Each daughter T cell is similarly activated and carries an identical TCR, thus amplifying the “knowledge” of antigen recognition.
各娘 T 細胞は同様に活性化され、同一の TCR を保持しているため、抗原認識の「知識」が増幅されます。
The activation and expansion of a CD4+ T cell response provide support for the activation and expansion of CD8+ T cells in the local lymph node.
CD4+ T 細胞応答の活性化と拡大は、局所リンパ節における CD8+ T 細胞の活性化と拡大をサポートします。
Co-stimulatory molecules (e.g., CD28 and 4-1BB) and cytokines, such as interleukin (IL)-12 and IL-2 secreted by innate immune cells and CD4+ T cells, enhance CD8+ T cell activation, prolonging division and stimulating specific transcriptional programs.
自然免疫細胞および CD4+ T 細胞によって分泌されるインターロイキン (IL)-12 および IL-2 などの共刺激分子 (CD28 や 4-1BB など) とサイトカインは、CD8+ T細胞の活性化を高め、分裂を延長し、特定の転写プログラムを刺激します。
As CD8+ T cells divide and differentiate, they express surface molecules, including ligands for death receptors to induce apoptosis in target infected cells, and also generate inflammatory cytokines and release intracellular stores of cytolytic molecules, including granzymes and perforin.
CD8+ T 細胞は分裂して分化するにつれて、標的感染細胞にアポトーシスを誘導するためのデスレセプターのリガンドなどの表面分子を発現し、炎症性サイトカインを生成し、グランザイムやパーフォリンなどの細胞溶解性分子の細胞内貯蔵物を放出します。
After their initial activation and expansion, chemokine gradients guide CD8+ T cells to traffic from the lymph node to the site of infection, where they ultimately execute effector function.
最初の活性化と増殖の後、ケモカイン勾配によって CD8+ T 細胞がリンパ節から感染部位へ移動するように誘導され、そこで最終的にエフェクター機能を実行します。
In the same lymph node, naive B cells also test their B cell receptors (BCRs) for binding viral antigens.
同じリンパ節では、ナイーブ B 細胞もウイルス抗原との結合について B 細胞受容体 (BCR) をテストします。
Rather than pMHC, BCRs recognize whole and intact proteins, and the BCR can bind to three-dimensional conformational epitopes on a viral protein’s surface.
BCR は pMHC ではなく、そっくりそのままのタンパク質を認識し、ウイルスタンパク質の表面上の三次元立体構造エピトープに結合できます。
Then, BCR-induced signaling, metabolic and transcriptional cascades, comparable to those induced by TCR signaling, activate B cells and initiate differentiation and replication.
次に、TCR シグナル伝達によって誘導されるものに匹敵する、BCR 誘導シグナル伝達、代謝および転写カスケードが B 細胞を活性化し、分化と複製を開始します。
Activated B cells and CD4+ T cells meet at the interface of T cell and B cell zones (T-B border), where BCR-activated B cells can present processed viral proteins as pMHC.
活性化されたB細胞とCD4+ T細胞は、T細胞とB細胞ゾーンの境界面(T-B境界)で出会い、そこでBCR活性化B細胞は、処理されたウイルスタンパク質をpMHCとして提示できます。
CD4+ T cells further promote B cell differentiation through additional ligand-receptor interactions (e.g., CD40L-CD40 and ICOS-ICOSL) and the secretion of cytokines (e.g., IL-4 and IL-21) that direct class switching to various isotypes (immunoglobulin [Ig]M, IgG, IgA, and IgE).
CD4+ T 細胞は、追加のリガンド-受容体相互作用 (例: CD40L-CD40 および ICOS-ICOSL) およびサイトカイン (例: IL-4 および IL-21) の分泌を通じて B 細胞の分化をさらに促進します。 さまざまなアイソタイプ (免疫グロブリン [Ig]M、IgG、IgA、および IgE) へのクラススイッチを指示します。
These interactions promote B cell differentiation into one of two divergent paths.
これらの相互作用は、B 細胞の 2 つの分岐経路のいずれかへの分化を促進します。
Some B cells can become short-lived antibody-secreting cells called plasmablasts, while others can enter specialized, organized structures called germinal centers.
一部の B 細胞は、形質芽細胞と呼ばれる短命の抗体分泌細胞になることができますが、その他の B 細胞は、胚中心と呼ばれる特殊な組織化された構造に入ることができます。
In germinal centers, B cells undergo a process called somatic hypermutation, where BCRs are diversified and their specificity altered through the introduction of mutations by the enzyme activation-induced cytidine deaminase (AID).
胚中心では、B 細胞は体細胞超突然変異と呼ばれるプロセスを受けます。このプロセスでは、酵素活性化誘導性シチジン デアミナーゼ (AID) による突然変異の導入によって BCR が多様化し、その特異性が変化します。
Newly mutated BCRs undergo a second round of competitive T-B cell interactions, referred to as selection, in which they must demonstrate their ability to bind and present antigen to specialized germinal center-localized CD4+ T cells.
新たに変異したBCRは、選択と呼ばれる第2ラウンドの競合的T-B細胞相互作用を受けます。 そこでは、胚中心に局在する特殊な CD4+ T 細胞に結合して抗原を提示する能力を実証する必要があります。
Depending on the timing and specific characteristics of the CD4+ T cell and B cell interactions in germinal centers, a second fate divergence occurs in which some “selected” B cells become memory B (Bmem) cells that are capable of responding to subsequent infections, while others become long-lived antibody-producing plasma cells that predominantly reside in the bone marrow.
胚中心における CD4+ T 細胞と B 細胞の相互作用のタイミングと特有の特性に応じて、一部の「選択された」B 細胞がその後の感染に応答できる記憶 B (Bmem) 細胞になるという第 2 の運命の分岐が発生します。 一方、主に骨髄に存在する長命の抗体産生形質細胞になる細胞もあります。
At full activation, a single plasma cell can produce and secrete thousands of antibodies per second.
完全に活性化すると、単一の形質細胞が毎秒数千の抗体を生成および分泌できます。
As this primary response matures, coordinated efforts by both localized and systemic lymphocytes and their synergistic effector functions control the pathogen.
この一次反応が成熟するにつれて、局所リンパ球と全身リンパ球の両方による協調的な取り組みとそれらの相乗的なエフェクター機能が病原体を制御します。
Following resolution of the infection, >90% of the effector CD4+ and CD8+ T cells, along with a substantial portion of short-lived plasmablast B cells, undergo a regulated process of apoptotic cell death (called contraction).
感染症の解決後、エフェクター CD4+ および CD8+ T 細胞の >90% が、短命の形質芽球 B 細胞のかなりの部分とともに、アポトーシス細胞死の制御されたプロセスを経ます。 (収縮と呼ばれます)。
After this contraction phase, there is still a relative increase in the number of antigen-specific B cells and T cells that can recognize the infection that just occurred (as compared with the naive compartment).
この収縮期の後でも、発生したばかりの感染を認識できる抗原特異的な B 細胞および T 細胞の数は (ナイーブ コンパートメントと比較して) 相対的に増加しています。
These cells form the reservoir of memory cells that can be quickly recalled on subsequent challenges, underlying immunity to re-infection.
これらの細胞は、再感染に対する免疫の基礎となる、その後の攻撃時にすぐに呼び戻される記憶細胞の貯蔵庫を形成します。
Antigen receptor recognition
抗原受容体認識
The critical recognition event for initiating an adaptive immune response is antigen receptor (TCR or BCR) binding to antigen (pMHC or intact protein epitopes, respectively).
適応免疫応答を開始するための重要な認識イベントは、抗原受容体 (TCR または BCR) が抗原 (それぞれ pMHC または無傷のタンパク質エピトープ) に結合することです。
How is it possible to seed the naive repertoire (the collection of antigen receptors within an organism) with enough diverse TCRs and BCRs to recognize various epitopes (the antigenic component recognized by the TCR or BCR), including those that are completely novel from an evolutionary perspective?
進化の観点から完全に新規のものを含むさまざまなエピトープ (TCR または BCR によって認識される抗原成分) を認識するのに十分な多様性のある TCR および BCR をナイーブ レパートリー (生物内の抗原受容体のコレクション) に播種することはどのようにして可能ですか?
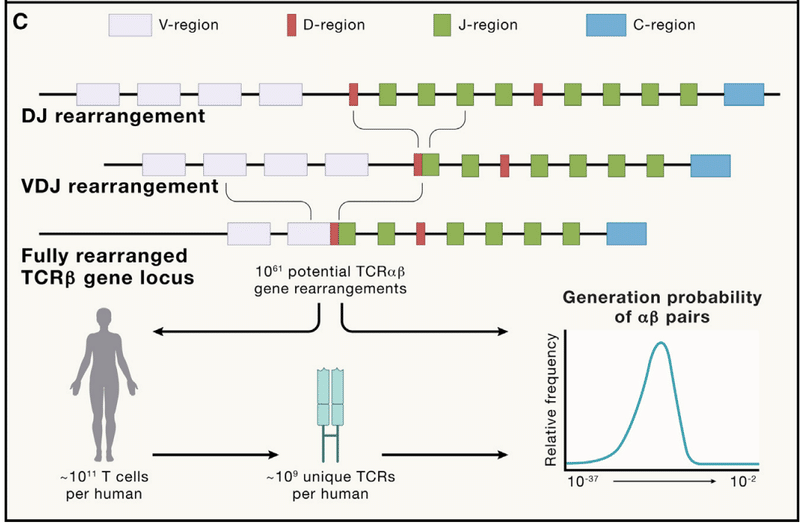
An extremely conservative estimate suggests that at least 10⁹ unique T cell clones exist in the naive repertoire (a number five orders of magnitude larger than the number of genes in the entire genome).
極めて控えめな推定では、ナイーブ レパートリーには少なくとも 10⁹ の固有の T 細胞クローンが存在すると示唆されています (この数は全ゲノムの遺伝子数より 5 桁多い)。
The solution to this limitation is that each T cell or B cell undergoes a highly coordinated process of somatic recombination to generate a unique antigen receptor with one heavy and one light chain.
この制限に対する解決策は、各 T 細胞または B 細胞が高度に調整された体細胞組み換えプロセスを受けて、1 つの重鎖と 1 つの軽鎖を持つ固有の抗原受容体を生成することです。
Mature lymphocytes change their genomes by re-arranging and ligating gene segments to generate novel coding sequences (Figure 1C).
成熟リンパ球は、遺伝子セグメントを再配置および連結して新しいコード配列を生成することによってゲノムを変化させます (図 1C)。
In humans, TCRs are either αβ or γδ (with the α and γ chains corresponding to the light chains).
ヒトでは、TCR は αβ または γδ のいずれかです (α 鎖と γ 鎖は軽鎖に対応します)。
For BCRs, the heavy chain is referred to as IgH, while the light chain is either Igκ or Igλ.
BCR の場合、重鎖は IgH と呼ばれ、軽鎖は Igκ または Igλ のいずれかです。
The TCR and BCR loci are extremely large (several hundred kilobases), with regions of multiple gene segment variants.
TCR および BCR 遺伝子座は非常に大きく (数百キロベース)、複数の遺伝子セグメント変異体の領域があります。
Each chain has both a cassette of “variable,” or V regions, and joining, or “J” regions, with the heavy chain containing an additional cassette of “diversity,” or D regions.
各鎖には「可変」または V 領域のカセットと結合または「J」領域の両方があり、重鎖には「多様性」または D 領域の追加カセットが含まれています。
To generate a receptor, the DNA is cut by the recombinase activating gene (RAG) enzymes and then stitched back together utilizing a specialized double-stranded break DNA repair process fusing one V, one D (for heavy chains), and one J region.
受容体を生成するには、DNA がリコンビナーゼ活性化遺伝子 (RAG) 酵素によって切断され、次に 1 つの V、1 つの D (重鎖用)、および 1 つの J 領域を融合する特殊な二本鎖切断 DNA 修復プロセスを利用してつなぎ合わされます。
DNA repair components Ku, DNA-dependent protein kinase (DNA-PK), Artemis, DNA ligase IV, and XRCC4 all participate in maintaining the proximity of the severed DNA strands and re-ligating them.
DNA 修復コンポーネント Ku、DNA 依存性プロテインキナーゼ (DNA-PK)、Artemis、DNA リガーゼ IV、および XRCC4 はすべて、切断された DNA 鎖の近接性の維持とそれらの再連結に関与しています。
In addition, terminal deoxynucleotidyl transferase (TdT) introduces template-independent nucleotides in the severed junction before ligation and repair, thereby providing an additional source of diversity in receptor generation to recognize a wide range of antigens.
さらに、ターミナル デオキシヌクレオチジル トランスフェラーゼ (TdT) は、ライゲーションと修復の前に切断された接合部にテンプレート非依存性ヌクレオチドを導入し、それによって広範囲の抗原を認識するための受容体生成における多様性のさらなる源を提供します。
Collectively, these sources of diversity (gene segment selection, combinatorial diversity, junctional diversity, and the pairing of heavy and light chains) result in an estimated 10^61 potentially unique TCRαβ chains.
これらの多様性の源(遺伝子セグメントの選択、組み合わせの多様性、接合の多様性、重鎖と軽鎖のペアリング)を総合すると、推定 10^61 個の潜在的にユニークな TCRαβ 鎖が生じます。
This number is exceptionally large—much higher than all of the T cells that have or will ever be generated in humans.
この数は非常に多く、これまでにヒトで生成された、または今後生成されるすべての T 細胞よりもはるかに多くなります。
Under conditions of random recombination, we would expect that no two T cells would arise within or between individuals with an identical receptor.
ランダムな組換えの条件下では、同一の受容体を持つ個体内または個体間で 2 つの T 細胞が発生しないと予想されます。
Although the occurrence of such “public” receptors is uncommon for the full two-chain combination, for single chains, sharing is observed in ∼15% of the TCRꞵ chains.
このような「パブリック」受容体の出現は、完全な 2 本鎖の組み合わせではまれですが、1 本鎖の場合、TCR 鎖の約 15% で共有が観察されます。
This observation indicates that the recombination process is strongly biased toward the generation of some receptor chains more than others.
この観察は、組換えプロセスが他の受容体鎖よりも一部の受容体鎖の生成に強く偏っていることを示しています。
A major accomplishment in the last several years, facilitated by the large amount of antigen receptor repertoire data produced by deep sequencing analyses, is the precise calculation of the probability of generating any given TCR or BCR.
ディープシークエンシング解析によって生成された大量の抗原受容体レパトアデータによって促進されたここ数年の主な成果は、特定の TCR または BCR が生成される確率の正確な計算です。
These approaches provide reliable estimates of how likely certain receptors are to occur, which can be verified by measuring the degree to which receptors are shared across the population.
これらのアプローチは、特定の受容体がどの程度発生する可能性があるかの信頼できる推定値を提供します。これは、受容体が集団全体でどの程度共有されているかを測定することで検証できます。
These tools have multiple applications, including for diagnostics and early detection of diseases, including chronic infections, tumors, and autoimmunity.
これらのツールには、慢性感染症、腫瘍、自己免疫などの病気の診断や早期検出など、複数の用途があります。
Beyond the intrinsically vast diversity available in the naive repertoire, B cells have a further mechanism for diversification of their antigen receptors.
ナイーブレパートリーで利用可能な本質的に広大な多様性を超えて、B 細胞は抗原受容体を多様化するためのさらなるメカニズムを持っています。
In the context of the germinal center structure described earlier, communication between CD4+ T cells and B cells leads to the induction of enzyme AID.
前述の胚中心構造の文脈では、CD4+ T 細胞と B 細胞の間のコミュニケーションにより、酵素 AID が誘導されます。
AID causes the deamination of cytidine, converting it to uracil in the DNA encoding the antigen receptor.
AID はシチジンの脱アミノ化を引き起こし、抗原受容体をコードする DNA 内でシチジンをウラシルに変換します。
The DNA repair pathway then converts this to a thymine or to other bases depending on the repair pathway engaged, thereby introducing a point mutation in the coding sequence.
次に、DNA修復経路は、関与する修復経路に応じてこれをチミンまたは他の塩基に変換し、それによってコード配列に点突然変異を導入します。
If this mutation enhances the affinity of the BCR for its ligand, the B cell will receive a survival signal.
この変異により BCR のリガンドに対する親和性が高まると、B 細胞は生存シグナルを受け取ることになります。
Otherwise, it will undergo apoptosis.
そうしないと、アポトーシスが発生します。
This process leads to the affinity maturation of the BCR (and the subsequent secreted antibodies), with the progression of the germinal center reaction resulting in a two-logarithm increase in the measured affinity of secreted antibodies over the course of a response.
このプロセスは、BCR (およびその後の分泌抗体) の親和性の成熟につながります。 胚中心反応の進行により、応答の過程で分泌された抗体の測定された親和性が 2 対数増加します。
The processes of V(D)J recombination and affinity maturation are intrinsically dangerous for the cell and the host, as they involve the purposeful breakage, re-ligation, and/or mutation of DNA.
V(D)J 組換えと親和性成熟のプロセスは、DNA の意図的な切断、再連結、および/または突然変異を伴うため、細胞と宿主にとって本質的に危険です。
Many pediatric and some adult tumors are driven by fusion genes created by illegitimate V(D)J recombination, demonstrating the embedded potential for malignant transformation in these processes.
多くの小児腫瘍と一部の成人腫瘍は、不正な V(D)J 組換えによって作成された融合遺伝子によって引き起こされており、これらのプロセスには悪性形質転換の可能性が埋め込まれていることが実証されています。
These include chronic myelogenous leukemia, which can contain a translocation between the BCR itself and Abl kinase, and T cell acute lymphoblastic leukemia, which can arise from RAG-mediated recombination between two non-antigen receptor genes, SIL and TAL1.
これらには、BCR自体とAblキナーゼの間の転座を含む可能性がある慢性骨髄性白血病や、2つの非抗原受容体遺伝子SILとTAL1の間のRAG媒介組換えから生じる可能性があるT細胞急性リンパ芽球性白血病が含まれます。
As a result, antigen receptor recombination is highly regulated by transcriptional induction (both of recombination machinery and at the recombination locus) and the physical sequestration of the antigen receptor loci within the nucleus in “recombination factories.”
その結果、抗原受容体の組換えは、転写誘導(組換え機構と組換え遺伝子座の両方)と、「組換え工場」における核内の抗原受容体遺伝子座の物理的隔離によって高度に制御されます。
There are many preventive and therapeutic strategies that could arise from harnessing this central means of immune recognition.
この免疫認識の中心的な手段を活用することで、多くの予防および治療戦略が生まれる可能性があります。
The ability to decode the repertoire is thus a focus of a number of investigators and is often called “the holy grail” of immunology, though it remains elusive.
したがって、レパートリーを解読する能力は多くの研究者の焦点であり、免疫学の「聖杯」と呼ばれることが多いが、依然としてとらえどころのないものである。
Although we cannot perform a simple mapping between the sequence of an antigen receptor and the identity of its target, significant progress has been made in extracting information from repertoire sequences.
抗原受容体の配列とその標的の正体との間の単純なマッピングを実行することはできませんが、レパートリー配列から情報を抽出することにおいては大きな進歩が見られました。
Receptors that recognize the same antigens often look very similar to each other.
同じ抗原を認識する受容体は、多くの場合、互いに非常によく似ています。
Finding identical paired chain receptors between individuals (“true public” receptors) is extremely unlikely, but receptors that share a high degree of homology are very common.
個人間で同一のペア鎖受容体 (「真の公共」受容体) が見つかる可能性は非常に低いですが、高度な相同性を共有する受容体は非常に一般的です。
These conserved motifs strongly suggest that the project of decoding the repertoire is likely to succeed, as it points to a conserved and limited set of solutions underlying how any particular antigen is seen by antigen receptors.
これらの保存されたモチーフは、特定の抗原が抗原受容体によってどのように見られるかの基礎となる、保存された限られた一連の解決策を示しているため、このレパートリーを解読するプロジェクトが成功する可能性が高いことを強く示唆しています。
Of note, only 17 epitopes with at least 50 unique paired TCRs are curated in the largest online database to date (accessed and analyzed in January 2024).
注目すべきことに、これまで最大のオンライン データベースで厳選されているのは、少なくとも 50 個の固有の TCR ペアを持つ 17 個のエピトープのみです (2024 年 1 月にアクセスおよび分析)。
Another complication for the prediction of specificity is the poorly understood extent of cross reactivity possible for any given receptor.
特異性の予測におけるもう 1 つの問題は、特定の受容体で起こり得る交差反応性の範囲が十分に理解されていないことです。
That receptors must be cross-reactive is known to be true.
受容体は交差反応性でなければならないということは真実であることが知られています。
Estimates as high as one million unique targets per TCR have been made, which imposes further challenges for the decoding problem.
TCR ごとに 100 万ものユニークターゲットが存在すると推定されており、デコード問題にさらなる課題が課せられています。
However, the translational potential of solving the repertoire code and recent advances in structural modeling keep this problem a major focus of current antigen receptor research.
しかし、レパートリーコードを解くトランスレーショナルの可能性と構造モデリングの最近の進歩により、この問題は現在の抗原受容体研究の主要な焦点となっています。
Balancing self versus non-self recognition: Positive and negative selection
自己認識と非自己認識のバランスをとる: ポジティブ選択とネガティブ選択
Considering the intrinsically stochastic nature of recombination, the generation of autoreactive TCRs and BCRs seems highly likely.
組換えの本質的な確率的性質を考慮すると、自己反応性 TCR および BCR が生成される可能性は非常に高いと思われます。
For TCRs, an additional constraint is that they need to recognize peptides in the context of MHC to function properly.
TCR の場合、追加の制約は、適切に機能するために MHC のコンテキストでペプチドを認識する必要があることです。
How then does T cell development ensure that such conditions (i.e., both lack of autoreactivity and restriction by MHC) are met?
では、T 細胞の発生はどのようにしてそのような条件 (すなわち、自己反応性の欠如と MHC による制限の両方) が満たされることを保証するのでしょうか?
For T cells, their development in the thymus includes a rigorous process of both positive and negative selection, often referred to as “thymic education.”
T 細胞の場合、胸腺での発達にはポジティブ選択とネガティブ選択の両方の厳密なプロセスが含まれます。 しばしば「胸腺教育」と呼ばれます。
Following recombination, receptors must bind to MHC to provide a survival signal; if this does not occur, the cell undergoes apoptosis, removing it from the population.
組換え後、受容体は MHC に結合して生存シグナルを発する必要があります;これが起こらない場合、細胞はアポトーシスを起こし、細胞集団から除去されます。
A receptor that binds MHC with too strong an affinity may induce apoptosis due to excessive activating signal, which helps eliminate self-reactive specificities from the repertoire (called central tolerance).
強すぎる親和性で MHC に結合する受容体は、過剰な活性化シグナルによりアポトーシスを誘発する可能性があり、これは自己反応性特異性をレパートリーから排除するのに役立ちます (中枢寛容と呼ばれます)。
To allow T cells to properly screen their receptors against antigens that will eventually be seen throughout the body, mechanisms exist to express diverse proteins from every major organ system and tissue in the thymus, with the transcription factor Aire, primarily expressed by medullary thymic epithelial cells, playing a central role in this process.
最終的に全身に現れる抗原に対する受容体を T 細胞が適切にスクリーニングできるようにするために、胸腺のあらゆる主要な器官系および組織から多様なタンパク質を発現するメカニズムが存在します。 転写因子 Aire は主に胸腺髄質上皮細胞によって発現され、このプロセスで中心的な役割を果たします。
Moreover, CD4+ T cells with moderate to high binding affinity for self-antigens are likely to upregulate expression of the transcriptional regulator Foxp3, which endows them the capacity to develop into a distinct lineage known as regulatory T (Treg) cells.
さらに、自己抗原に対して中程度から高い結合親和性を持つ CD4+ T 細胞は、転写調節因子 Foxp3 の発現を上方制御する可能性があります。 これにより、制御性 T (Treg) 細胞として知られる異なる系統に発達する能力が与えられます。
After exiting the thymus, Treg cells mediate crucial immunosuppressive function to prevent autoimmunity but also have emerging roles in supporting tissue repair and physiology, and these two effects are described further below.
胸腺を出た後、Treg 細胞は自己免疫を防ぐために重要な免疫抑制機能を仲介しますが、組織修復と生理機能をサポートする新たな役割も担っており、これら 2 つの効果については以下でさらに説明します。
Developing B cells also undergo processes of selection while they are developing in the bone marrow.
発生中の B 細胞は、骨髄で発生する間に選択のプロセスも受けます。
Self-reactive, immature B cells can be deleted, or alternatively, their BCRs can undergo “receptor editing,” which allows for additional gene recombination events to produce a non-self-reactive BCR.
自己反応性の未熟な B 細胞を削除することも、その BCR に「受容体編集」を施すこともできます。これにより、追加の遺伝子組み換えイベントが可能になり、非自己反応性 BCR が生成されます。
Altogether, given the potentially deleterious effects of adaptive immune responses to host tissues, these developmental processes are crucial for “educating” T cells and B cells by equipping them with proper antigen receptors to mediate productive immune responses while avoiding self-disruption.
まとめると、宿主組織に対する適応免疫応答の潜在的に有害な影響を考慮すると、 これらの発生プロセスは、自己破壊を回避しながら生産的な免疫応答を媒介する適切な抗原受容体を T 細胞と B 細胞に備えさせることで、T 細胞と B 細胞を「教育」するために重要です。
What kind of responses does adaptive immunity generate in the periphery?
適応免疫は末梢でどのような反応を引き起こすのでしょうか?
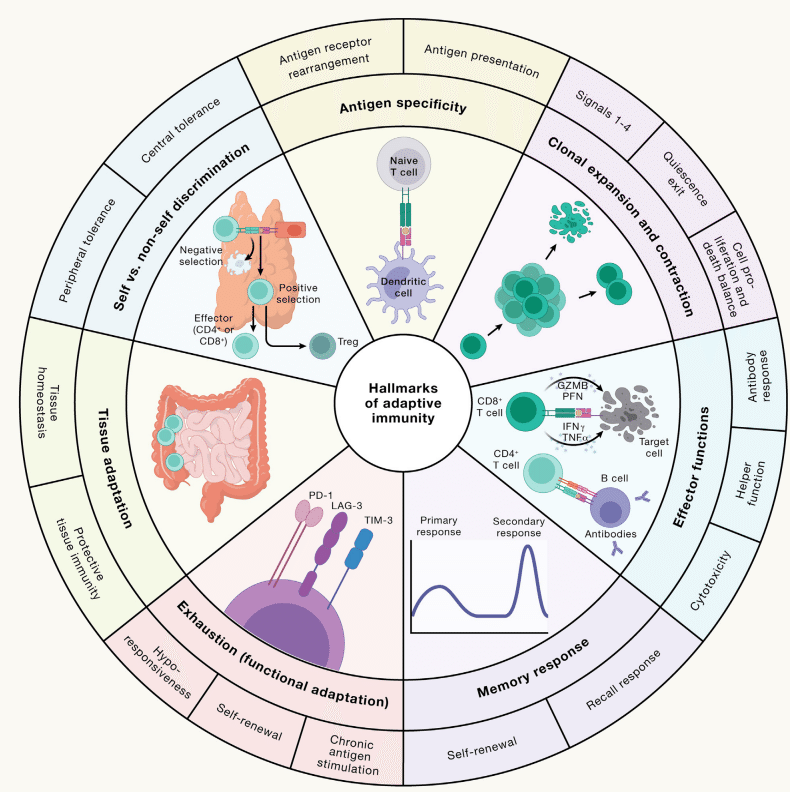
Appropriate activation of adaptive immunity requires reprogramming of naive lymphocytes into effector cells, which are crucial to defend the host against invading pathogens or to influence other immune or tissue cells (Figure 2).
適応免疫を適切に活性化するには、ナイーブリンパ球をエフェクター細胞に再プログラミングする必要があります。エフェクター細胞は、侵入する病原体から宿主を防御したり、他の免疫細胞や組織細胞に影響を与えるために重要です(図2)。
How does the adaptive immune system in the peripheral lymphoid organs (primarily consisting of spleen and lymph nodes) respond to diverse threats initially, re-encounter them after initial clearance, or adapt to constantly facing them?
末梢リンパ器官 (主に脾臓とリンパ節からなる) の適応免疫系は、最初はさまざまな脅威にどのように反応しますか? 最初のクリア後に彼らに再び遭遇しますか、それとも常に彼らと対峙することに適応しますか?
Also, what mechanisms prevent adaptive immunity from carrying out autoimmune reactions in response to self-antigens expressed in the peripheral tissues (called peripheral tolerance)?
また、末梢組織で発現される自己抗原に応答して適応免疫が自己免疫反応を実行するのを妨げるメカニズムは何ですか (末梢寛容と呼ばれます)。
We discuss effector and memory responses, T cell functional adaptation to chronic antigen stimulation, and Treg-mediated peripheral tolerance and restriction of inflammation (Figure 3), which collectively balance protective immunity against autoimmune reactions.
私たちは、エフェクターと記憶の応答、慢性抗原刺激に対する T 細胞の機能的適応、および Treg を介した末梢寛容と炎症の制限 (図 3) について説明します。これらは集合的に自己免疫反応に対する防御免疫のバランスをとります。
Diverse effector responses: Responding to different types of threats
多様なエフェクターの応答: さまざまな種類の脅威に対応
How does the immune system respond to the vast diversity of invading pathogens and immunological insults?
免疫系は、多種多様な侵入病原体や免疫学的損傷にどのように反応するのでしょうか?
One solution is the generation of specialized effector cells that display functional diversity based on signals derived from the priming APCs and changing environments.
解決策の 1 つは、プライミング APC および変化する環境から得られるシグナルに基づいて機能的多様性を示す特殊化されたエフェクター細胞の生成です。
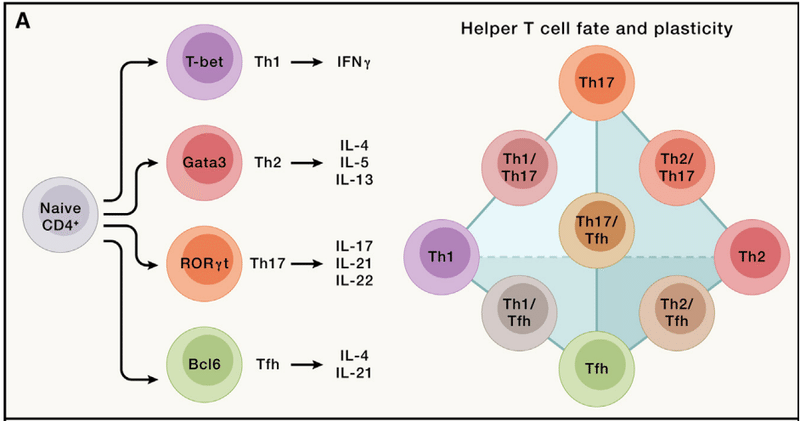
For example, CD4+ T cells respond to pro-inflammatory cues and differentiate into effector cells, including the Th1, Th2, and Th17 subsets (“h” stands for “helper”; namely, the function to help other immune cells such as CD8+ T cells or B cells) of cells that mediate protective immune responses following bacterial, helminth, and fungal infections, respectively.
たとえば、CD4+ T 細胞は炎症誘発性の合図に応答し、エフェクター細胞に分化します。 これには、それぞれ細菌、蠕虫、真菌感染後の防御免疫応答を媒介する細胞の Th1、Th2、および Th17 サブセットが含まれます(「h」は「ヘルパー」の略で、CD8+ T細胞やB細胞など他の免疫細胞を助ける機能)。
These functions are orchestrated by lineage-specific transcription factors (T-bet, Gata3, and RORγt), signature cytokines, and other effector molecules.
これらの機能は、系統特異的な転写因子 (T-bet、Gata3、RORγt)、特徴的なサイトカイン、およびその他のエフェクター分子によって調整されます。
In addition, Bcl6-expressing CD4+ T follicular helper (Tfh) cells are crucial for supporting B cell-mediated antibody responses (Figure 3A).
さらに、Bcl6 発現 CD4+ T 濾胞ヘルパー (Tfh) 細胞は、B 細胞媒介抗体応答をサポートするために重要です (図 3A)。
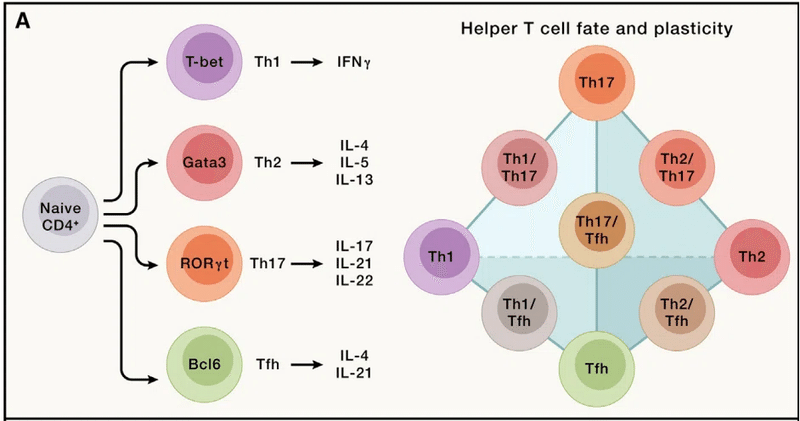
However, effector cells are not simply fixed lineages, and they adapt their functions to ever-changing microenvironmental and immunological cues, a phenomenon called plasticity.
しかし、エフェクター細胞は単に固定された系統ではなく、常に変化する微小環境や免疫学的手がかりにその機能を適応させます(可塑性と呼ばれる現象)。
Of note, Th17 cells display more plasticity than Th1 or Th2 cells.
注目すべきことに、Th17 細胞は Th1 または Th2 細胞よりも高い可塑性を示します。
For instance, Th17 cells can adopt IL-23-mediated Th1-like features with enhanced capacity to induce autoimmunity and inflammation, and mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1)-dependent metabolic rewiring contributes to this process.
たとえば、Th17 細胞は、自己免疫と炎症を誘導する能力が強化された、IL-23 媒介の Th1 様の特徴を採用できます。 そして、ラパマイシン複合体 1 (mTORC1) の機構的 (または哺乳類) 標的依存性代謝再配線がこのプロセスに寄与しています。
Th17 cells may also transdifferentiate into IL-10-producing cells (Tr1) with immunosuppressive function to resolve inflammation.
Th17 細胞は、炎症を解消するために免疫抑制機能を持つ IL-10 産生細胞 (Tr1) に分化転換することもあります。
Further, Th17 cells can produce IL-4 in helminth infection and possibly allergic asthma.
さらに、Th17 細胞は、蠕虫感染症やおそらくアレルギー性喘息において IL-4 を産生する可能性があります。
Beyond plasticity, effector responses are functionally heterogeneous.
可塑性を超えて、エフェクター応答は機能的に不均一です。
For example, homeostatic (non-pathogenic) Th17 cells promote tissue homeostasis and prevention of microbial infection under steady state, especially at mucosal sites such as the intestine, whereas pathogenic Th17 cells precipitate inflammatory and autoimmune diseases.
たとえば、恒常性のある(非病原性)Th17 細胞は、定常状態下、特に腸などの粘膜部位で組織の恒常性と微生物感染の予防を促進しますが、病原性 Th17 細胞は炎症性疾患や自己免疫疾患を引き起こします。
Th17 cell pathogenicity is associated with co-expression of IFNγ and granulocyte-macrophage colony-stimulating factor (GM-CSF) and requires rewiring of glucose, lipid, and polyamine metabolism.
Th17 細胞の病原性は、IFNγ と顆粒球マクロファージ コロニー刺激因子 (GM-CSF) の共発現に関連しており、グルコース、脂質、およびポリアミン代謝の再配線を必要とします。
Moreover, Tfh cell function is tailored in response to specific immunological insults, which supports class switching to various antibody isotypes that provide protective immunity to different types of pathogens.
さらに、Tfh 細胞の機能は特定の免疫学的損傷に応答して調整されており、さまざまな種類の病原体に対する防御免疫を提供するさまざまな抗体アイソタイプへのクラススイッチをサポートします。
The inherent plasticity and heterogeneity of CD4+ T cells for functionally adapting to changing environments may be harnessed for therapeutic interventions.
変化する環境に機能的に適応するための CD4+ T 細胞の固有の可塑性と不均一性は、治療介入に利用される可能性があります。
Activated B cells proliferate and migrate to the T-B border, where they interact with Tfh cells.
活性化された B 細胞は増殖して T-B 境界に移動し、そこで Tfh 細胞と相互作用します。
Tfh cells provide multiple helper signals, including cytokines that direct immunoglobulin class switching (from IgD and IgM to IgG, IgE, or IgA isotypes), resulting in a pool of B cells with diversified functions.
Tfh 細胞は、免疫グロブリンのクラススイッチ (IgD および IgM から IgG、IgE、または IgA アイソタイプへ) を指示するサイトカインを含む複数のヘルパー シグナルを提供し、その結果、多様な機能を持つ B 細胞のプールが形成されます。
Recent high-dimensional profiling, especially single-cell RNA sequencing (scRNA-seq), has revealed additional heterogeneity of T cells, B cells, and other adaptive immune cells, including in patient populations with cancer, rheumatoid arthritis, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, and neurodegenerative diseases.
最近の高次元プロファイリング、特に単一細胞 RNA シーケンス (scRNA-seq) により、T 細胞、B 細胞、およびその他の適応免疫細胞の不均一性が明らかになりました。 これには、がん、関節リウマチ、重症急性呼吸器症候群コロナウイルス 2 (SARS-CoV-2) 感染症、神経変性疾患の患者集団が含まれます。
For instance, scRNA-seq of patients with bladder cancer revealed the existence of clonally expanded cytotoxic CD4+ T cell states.
例えば、膀胱癌患者の scRNA-seq により、クローン的に増殖した細胞傷害性 CD4+ T 細胞状態の存在が明らかになりました。
These cells have the capacity for killing autologous tumor cells, and their gene signature predicts response to anti-PD-L1 treatment, further supporting the notion of diversity and heterogeneity of adaptive immune responses.
これらの細胞は自己腫瘍細胞を殺す能力があり、その遺伝子サインは抗 PD-L1 治療に対する反応を予測します。 適応免疫応答の多様性と不均一性の概念をさらに裏付けるものです。
Immune memory: Remembering the threats upon re-encounter
免疫記憶: 脅威に再び遭遇したときに思い出す
Immune memory, a hallmark of adaptive immunity (Figure 2), enables a rapid and enhanced immune response upon antigen re-encounter.
適応免疫の特徴である免疫記憶 (図 2) により、抗原との再遭遇時に迅速かつ強化された免疫応答が可能になります。
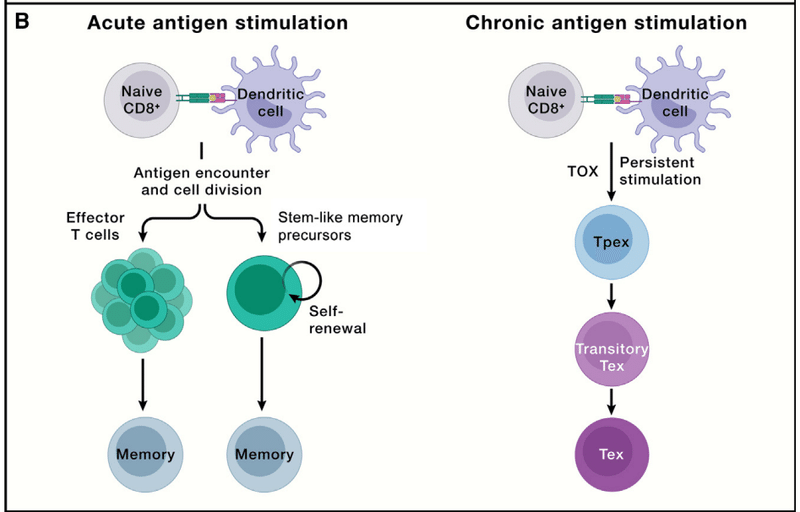
This effect is associated with an increased frequency of antigen-specific T cells with intrinsically enhanced functionality compared with naive populations and is studied in all lymphocytes, especially CD8+ T cells (Figure 3B).
この効果は、ナイーブ集団と比較して、本質的に機能が強化された抗原特異的 T 細胞の頻度の増加に関連しており、すべてのリンパ球、特に CD8+ T 細胞で研究されています (図 3B)。
Whereas the majority of antigen-specific effector T cells undergo contraction, long-lived CD8+ T cells may descend from a subset of effector cells that retain the capacity to re-acquire stem-like characteristics (i.e., signature molecules expressed by naive T cells).
抗原特異的エフェクター T 細胞の大部分は収縮しますが、長命の CD8+ T 細胞は、幹様の特徴を再獲得する能力を保持しているエフェクター細胞のサブセット (つまり、ナイーブ T 細胞によって発現されるシグネチャー分子)に由来している可能性があります。
This effector to memory cell transition is a “de-differentiation” process orchestrated by epigenetic mechanisms like DNA demethylation.
このエフェクターから記憶細胞への移行は、DNA 脱メチル化などのエピジェネティックなメカニズムによって調整される「脱分化」プロセスです。
Alternatively, memory cells may emerge from a self-renewing population before or during the clonal expansion phase following antigen stimulation, thereby linking differentiation to replicative history.
あるいは、記憶細胞は、抗原刺激後のクローン増殖期の前または最中に自己再生集団から出現し、それによって分化を複製履歴に結び付ける可能性があります。
Further, asymmetric partitioning of key signaling molecules during the first cell division may reinforce memory formation.
さらに、最初の細胞分裂中の重要なシグナル伝達分子の非対称な分割が記憶形成を強化する可能性があります。
Transcriptional and metabolic pathways control memory CD8+ T cell differentiation, which require TCF-1, Myb, and Foxo1, as well as metabolic processes (e.g., mitochondrial oxidative phosphorylation [OXPHOS] and mitochondrial dynamics).
転写経路および代謝経路は、記憶 CD8+ T 細胞の分化を制御します。これには、TCF-1、Myb、および Foxo1、ならびに代謝プロセス (例、ミトコンドリアの酸化的リン酸化 [OXPHOS] およびミトコンドリアの動態) が必要です。
Future studies using cutting-edge lineage-tracing tools are needed to reconcile or unify these differentiation models and identify the underlying mechanisms.
これらの分化モデルを調整または統合し、根底にあるメカニズムを特定するには、最先端の系統追跡ツールを使用した今後の研究が必要です。
For an effective recall response to occur upon pathogen rechallenge, memory CD8+ T cells found at the site of infection, in lymphoid organs, and in circulation all participate, with each subset exhibiting differential capabilities for self-renewal, longevity, and cytotoxic function.
病原体再攻撃時に効果的な想起反応が起こるためには、感染部位、リンパ器官、循環器に存在するメモリー CD8+ T 細胞がすべて関与しています。 各サブセットは、自己再生、寿命、細胞毒性機能に関して異なる能力を示します。
The memory pool mainly includes central memory (TCM), effector memory (TEM), and tissue-resident memory (TRM) cells, although stem cell-like memory (TSCM) and effector memory re-expressing CD45RA (TEMRA) cells have also been described.
メモリ プールには主に、セントラル メモリ (TCM)、エフェクター メモリ (TEM)、および組織常駐メモリ (TRM) 細胞が含まれます。 ただし、幹細胞様記憶 (TSCM) 細胞やエフェクター記憶再発現 CD45RA (TEMRA) 細胞も報告されています。
TCM cells are quiescent memory cells expressing low levels of cytotoxic markers and high levels of lymphoid-tissue homing markers (e.g., CCR7 and CD62L) for their localization to lymphoid tissues.
TCM 細胞は、リンパ組織への局在化のために、低レベルの細胞傷害性マーカーと高レベルのリンパ組織ホーミング マーカー (CCR7 や CD62L など) を発現する静止記憶細胞です。
TCM cells may undergo self-renewal in response to IL-7 and IL-15 and differentiate into TEM cells.
TCM 細胞は、IL-7 および IL-15 に応答して自己複製を起こし、TEM 細胞に分化する可能性があります。
By contrast, TEM cells display intermediate longevity, express lower levels of these homing markers, and circulate between the blood and inflamed non-lymphoid tissues.
対照的に、TEM 細胞は中程度の寿命を示し、これらのホーミング マーカーの発現レベルは低く、血液と炎症を起こした非リンパ組織の間を循環します。
Upon re-infection, TEM cells are rapidly recruited from circulation to provide immune defense due to their inherent cytotoxic function.
再感染すると、TEM 細胞はその固有の細胞毒性機能により免疫防御を提供するために循環から急速に補充されます。
Germinal centers support the generation of the memory B cell pool, which forms after an iterative process of BCR diversification and affinity maturation.
胚中心は、BCR の多様化と親和性成熟の反復プロセスの後に形成される記憶 B 細胞プールの生成をサポートします。
Two different memory B cell populations emerge: long-lived antibody-secreting plasma cells and Bmem cells.
長寿命の抗体分泌形質細胞と Bmem 細胞という 2 つの異なる記憶 B 細胞集団が出現します。
Long-lived plasma cells produce serum antibodies that can neutralize a pathogen and prevent infection.
長命の形質細胞は、病原体を中和して感染を防ぐことができる血清抗体を生成します。
Bmem cells are rapidly re-activated upon subsequent antigen re-encounter and BCR engagement, causing them to proliferate and rapidly produce expanded populations of antibody-secreting cells.
Bmem 細胞は、その後の抗原の再遭遇と BCR の結合により急速に再活性化されます。 これにより、それらが増殖し、急速に拡大された抗体分泌細胞の集団が生成されます。
Bmem cells circulate and survey the body for their antigen (or a closely related variant), or alternatively take residence in tissues to protect against re-infection.
Bmem 細胞は体内を循環し、その抗原 (または密接に関連した変異体) を探します。 あるいは、再感染を防ぐために組織内に定着します。
Thus, diversified T cell and B cell subsets are involved in establishing immune memory.
したがって、多様な T 細胞および B 細胞サブセットが免疫記憶の確立に関与しています。
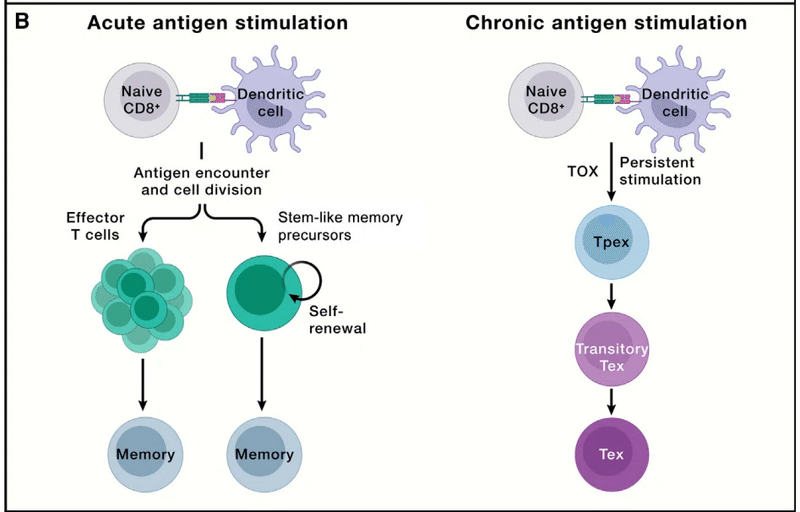
T cell exhaustion: Facing the threats all the time
T細胞の枯渇: 常に脅威に直面している
Although effector lymphocytes function to clear invading pathogens in acute infection, chronic infection, such as HIV, occurs when pathogens are not resolved, leading to persistent antigen stimulation.
エフェクターリンパ球は急性感染症において侵入した病原体を除去する機能を持っていますが、 HIV などの慢性感染症において病原体が解決されない場合に持続的な抗原刺激につながる事態が発生します。
Under such conditions, T cells gradually become functionally exhausted, a state associated with dampened effector function, which is also observed in progressive tumors (Figure 3B).
このような条件下では、T 細胞は徐々に機能的に疲弊し、エフェクター機能の低下に関連する状態になります。これは進行性腫瘍でも観察されます (図 3B)。
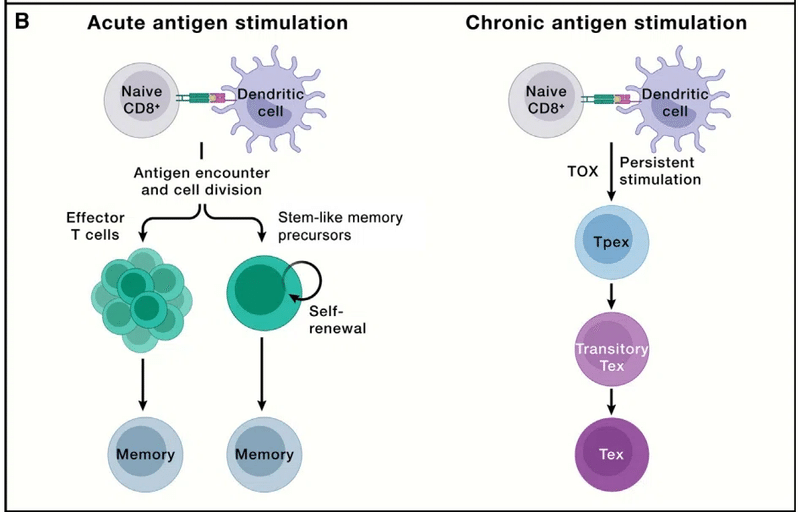
T cell exhaustion prevents tissue damage and excessive immunopathology while also contributing to the curtailment of pathogen and tumor growth, thereby reflecting a hypofunctional state of adaptation.
T細胞の枯渇は、組織の損傷や過剰な免疫病理を防止すると同時に、病原体や腫瘍の増殖の抑制にも貢献します。 したがって、適応の機能低下状態を反映しています。
Exhausted CD8+ T cells upregulate the expression of co-inhibitory molecules (namely, PD-1, LAG3, TIM-3, and TIGIT); gradually reduce proliferation; and display a unique chromatin landscape compared with effector and memory T cells.
枯渇CD8+ T 細胞は、共抑制分子 (つまり、PD-1、LAG3、TIM-3、および TIGIT) の発現を上方制御します;徐々に増殖を減少;エフェクター T 細胞やメモリー T 細胞と比較して、独特のクロマチンランドスケープを示します。
TOX is the master transcriptional regulator for inducing the exhaustion program but is largely dispensable for effector or memory T cell formation during acute infection.
TOX は枯渇プログラムを誘導するマスター転写制御因子ですが、急性感染時のエフェクターまたはメモリー T 細胞の形成にはほとんど必要ありません。
Given the functions of CD8+ T cells in mediating adaptive immunity to tumors and chronic infection, the mechanisms underlying T cell exhaustion and how to reinvigorate their functionality are being actively investigated.
腫瘍や慢性感染症に対する適応免疫の媒介における CD8+ T 細胞の機能を考慮すると、T 細胞枯渇の根底にあるメカニズムとその機能を再活性化する方法が積極的に研究されています。
These efforts led to the discovery that exhausted T cells are separated into two major functional subsets: TCF-1+ precursor exhausted (Tpex) and TCF-1− terminally exhausted T (Tex) cells (Figure 3B).
これらの取り組みにより、枯渇した T 細胞が 2 つの主要な機能サブセットに分けられることが発見されました。 TCF-1+ 前駆体枯渇 (Tpex) 細胞と TCF-1- 最終枯渇 T (Tex) 細胞 (図 3B)。
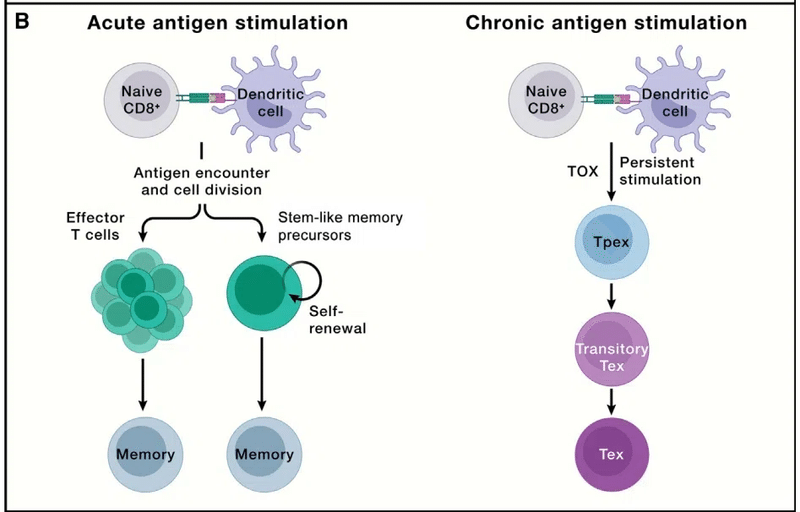
Tpex cells exhibit inherent stemness and self-renewal capacity and directly respond to antibody-based immunotherapies (called immune checkpoint blockade [ICB]) by producing a proliferative burst of functional CD8+ T cells to control pathogens or tumors.
Tpex 細胞は固有の幹細胞性と自己再生能力を示し、病原体や腫瘍を制御するために機能的な CD8+ T 細胞の増殖バーストを生成することにより、抗体ベースの免疫療法 (免疫チェックポイント阻害 [ICB] と呼ばれます) に直接応答します。
Among Tpex cells, a transcriptionally distinct CD62L+ Tpex cell population retains long-term proliferative potential and multipotency in a Myb-dependent manner.
Tpex 細胞の中で、転写的に異なる CD62L+ Tpex 細胞集団は、Myb 依存的に長期の増殖能と多分化能を保持します。
Transitory exhausted CD8+ T cells (with heightened effector function) are an intermediate state between Tpex and Tex cells.
一時的枯渇CD8+ T 細胞 (エフェクター機能が高まった) は、Tpex 細胞と Tex 細胞の間の中間状態です。
Further, transcriptional regulomes program the stepwise differentiation of exhausted CD8+ T cells, and enforcing Tpex cell quiescence exit and transitory Tex cell accumulation both bolster antitumor effects.
さらに、転写レギュロームは枯渇 CD8+ T 細胞の段階的な分化をプログラムし、Tpex 細胞の静止離脱と一時的な Tex 細胞の蓄積の両方を強制して抗腫瘍効果を強化します。
Of note, antigen-specific T cells may also undergo terminal exhaustion without progression through a functional effector state.
注目すべきことに、抗原特異的 T 細胞は、機能的なエフェクター状態を経ずに最終的に枯渇することもあります。
The differentiation trajectory of exhausted T cells in discrete chronic infections or tumors warrants further investigation.
個別の慢性感染症または腫瘍における枯渇 T 細胞の分化軌跡については、さらなる研究が必要です。
Spatiotemporal control of CD8+ T cell responses is evident in tumors.
CD8+ T 細胞応答の時空間制御は腫瘍において明らかです。
Tumor-draining lymph nodes (tdLNs) contain an abundant Tpex cell population (possibly derived from tumor-specific memory (TTSM) cells), which requires MHC-I-dependent interaction with type 1 conventional DCs (cDC1s) in the T cell and marginal zones of tdLNs for their maintenance.
腫瘍流入リンパ節 (tdLN) には、豊富な Tpex 細胞集団 (おそらく腫瘍特異的記憶 (TTSM) 細胞に由来する) が含まれています。これには、その維持のために、T細胞およびtdLNの周縁領域における1型従来型DC(cDC1)とのMHC-I依存性相互作用が必要である。
When Tpex cells migrate from tdLNs to the tumor, they undergo terminal differentiation.
Tpex 細胞が tdLN から腫瘍に移動すると、最終分化が起こります。
Further, the tertiary lymphoid structure facilitates the influx of B cells and T cells into the tumor site, which has been shown to support distinct subsets of exhausted T cells.
さらに、三次リンパ構造は腫瘍部位への B 細胞と T 細胞の流入を促進し、枯渇 T 細胞の異なるサブセットをサポートすることが示されています。
Applications of spatial transcriptomics technologies and lineage tracing tools will help decipher cell state transitions and cell-cell interactions underlying lymphocyte adaptation across space and time.
空間トランスクリプトミクス技術と系統追跡ツールの応用は、時空を超えたリンパ球の適応の根底にある細胞状態遷移と細胞間相互作用を解読するのに役立ちます。
Treg cells limit autoimmunity and inflammation
Treg細胞は自己免疫と炎症を制限する
A defining feature of the adaptive immune system is the ability to distinguish self-tissue antigens from “non-self” (e.g., invading pathogens) (Figure 2).
適応免疫系の決定的な特徴は、自己組織抗原を「非自己」抗原(侵入病原体など)から区別する能力です(図 2)。
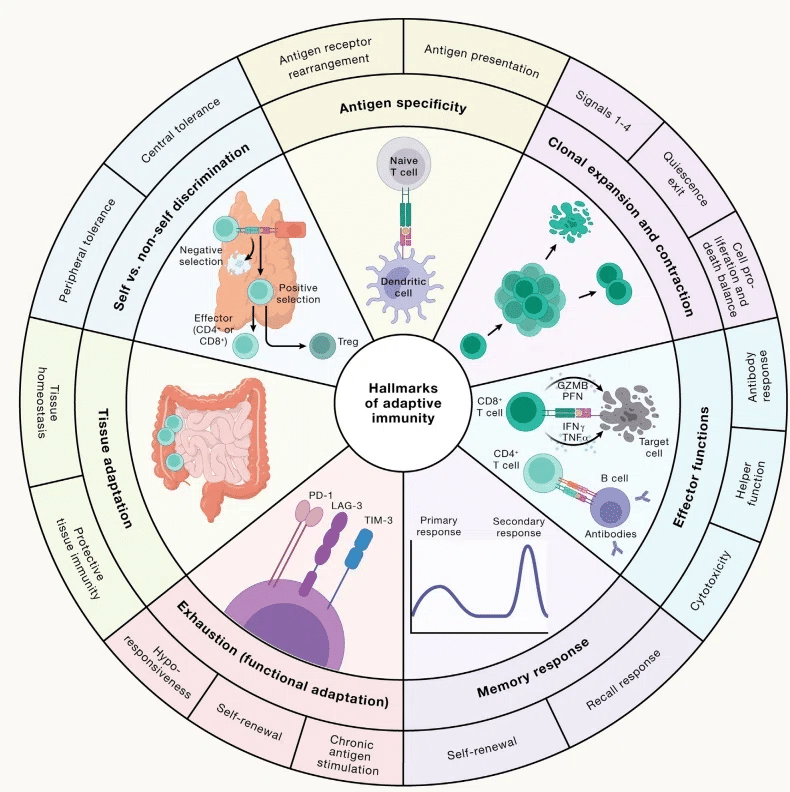
Although central tolerance is crucial for immune homeostasis, it is only partially effective.
中枢性寛容は免疫恒常性にとって極めて重要ですが、効果は部分的にしかありません。
Therefore, mechanisms of peripheral tolerance exist to prevent autoimmune reactions, including cell-intrinsic mechanisms (e.g., lymphocyte quiescence, ignorance, anergy, and senescence, as reviewed elsewhere) and extrinsic control, predominantly mediated by Treg cells.
したがって、末梢寛容のメカニズムは、細胞固有のメカニズム(例えば、他の場所でレビューされているように、リンパ球の静止、免疫学的無知、アネルギー、および老化)および主にTreg細胞によって媒介される外因性制御を含む自己免疫反応を防ぐために存在します。
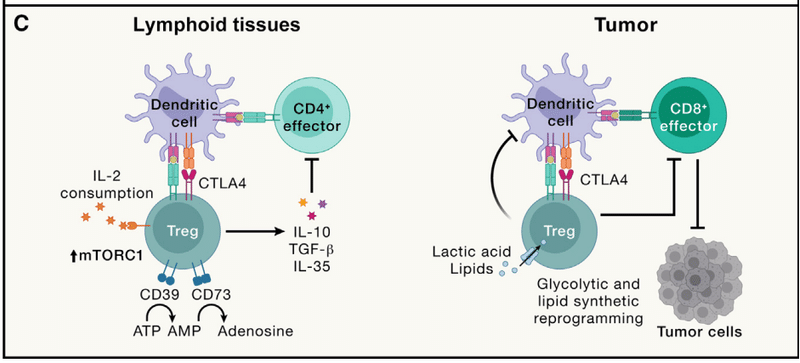
Loss of these cells or their lineage-defining transcription factor Foxp3 triggers early-onset fatal autoimmunity in mice and humans, suggesting that Treg cells act as a cellular “brake” to prevent adaptive immunity from targeting self-tissue (Figure 3C).
これらの細胞またはその系統を決定する転写因子 Foxp3 の喪失は、マウスやヒトにおいて早期に致死的な自己免疫を引き起こし、Treg 細胞が自己組織を標的とした適応免疫を防ぐ細胞の「ブレーキ」として機能することを示唆しています (図 3C)。
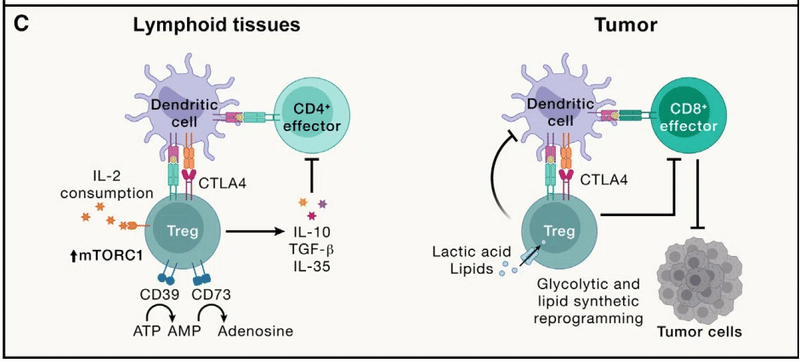
Treg cells require Foxp3 for their function and also adapt to enforce context-specific immune regulation mediated by immunosuppressive cytokines (e.g., IL-10, TGF-β, and IL-35); co-inhibitory molecule CTLA4; and ectonucleotidases CD39 and CD73 to deplete pro-inflammatory extracellular ATP.
Treg 細胞はその機能に Foxp3 を必要とし、免疫抑制性サイトカイン (IL-10、TGF-β、IL-35 など) によって媒介される状況特異的な免疫制御を強制するようにも適応します;共阻害分子CTLA4;炎症促進性細胞外ATPを枯渇させるエクトヌクレオチダーゼCD39およびCD73。
These diverse mechanisms contribute to the immunosuppressive functions of Treg cells in self and tumor tolerance.
これらの多様なメカニズムは、自己寛容および腫瘍寛容における Treg 細胞の免疫抑制機能に寄与しています。
To balance effector responses, Treg cells undergo adaptation in the periphery.
エフェクター応答のバランスをとるために、Treg 細胞は末梢で適応を受けます。
Compared with naive T cells, Treg cells display an antigen-experienced, activated phenotype due to their TCR-mediated recognition of self-tissue and also require IL-2 signaling and mTORC1-mediated anabolic metabolism for functional fitness.
ナイーブ T 細胞と比較して、Treg 細胞は、TCR を介した自己組織の認識により、抗原を経験した活性化された表現型を示し、機能的フィットネスのために IL-2 シグナル伝達と mTORC1 を介した同化代謝も必要とします。
Further, in response to specific cytokine and inflammatory cues, Treg cells co-opt the transcriptional and trafficking programs of effector T cells, resembling plastic effector responses (Figure 3A).
さらに、特定のサイトカインや炎症の合図に応答して、Treg 細胞はエフェクター T 細胞の転写および輸送プログラムを利用します。可塑性エフェクター応答に似ています (図 3A)。
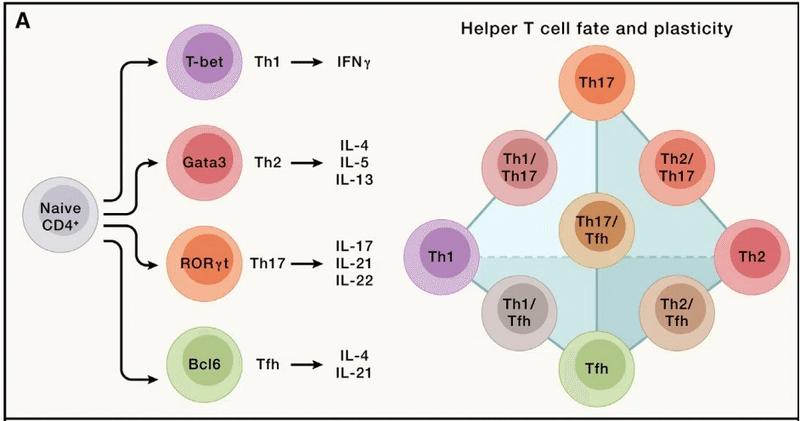
For instance, under Th1-prone conditions, Treg cells acquire T-bet and CXCR3 expression and Th1-like programming to counteract T-bet-expressing CD4+ and CD8+ T cells.
例えば、Th1傾向の条件下では、Treg細胞はT-betおよびCXCR3の発現とTh1様プログラミングを獲得して、T-betを発現するCD4+およびCD8+ T細胞に対抗します。
Further, follicular Treg cells express Bcl6 and CXCR5 to downmodulate Tfh cell activity and germinal center reactions.
さらに、濾胞性 Treg 細胞は Bcl6 と CXCR5 を発現して、Tfh 細胞活性と胚中心反応を下方調節します。
The plasticity of Treg cells ensures a precise spatiotemporal control of the context-specific effector responses while limiting immunopathology.
Treg 細胞の可塑性により、免疫病理を制限しながら、状況特異的なエフェクター応答の正確な時空間制御が保証されます。
Although Treg cells are beneficial for mediating protection from autoimmunity, restraining effector responses may be detrimental in certain conditions.
Treg 細胞は自己免疫からの防御を仲介するのに有益ですが、エフェクター応答の抑制は特定の状態では有害となる可能性があります。
In particular, intratumoral Treg cell accumulation is a key barrier to antitumor immunity and immunotherapy and correlates with decreased patient survival (Figure 3C).
特に、腫瘍内 Treg 細胞の蓄積は、抗腫瘍免疫および免疫療法に対する重要な障壁であり、患者の生存率の低下と相関しています (図 3C)。
Intratumoral Treg cells are heterogeneous, and only the FOXP3hi, but not FOXP3lo, subpopulation shows immunosuppressive capabilities in patients with colorectal cancer.
腫瘍内 Treg 細胞は不均一であり、 また、結腸直腸がん患者において免疫抑制能を示すのは FOXP3lo ではなく、FOXP3hi の亜集団のみです。
Additionally, Th1-like Treg cells accumulate in tumors and co-localize with CXCL9-producing cDC1s to suppress CD8+ T cell function.
さらに、Th1 様 Treg 細胞は腫瘍内に蓄積し、CXCL9 産生 cDC1 と共局在して CD8+ T 細胞の機能を抑制します。
However, intratumoral Treg cell adaptation can result in aberrant IFNγ expression that reprograms the tumor microenvironment to be more inflammatory, thereby boosting antitumor immunity.
しかし、腫瘍内 Treg 細胞の適応により、異常な IFNγ 発現が引き起こされ、腫瘍微小環境がより炎症性になるように再プログラムされ、それによって抗腫瘍免疫が増強されることがあります。
腫瘍内 Treg 細胞は、乳酸や脂質などの栄養素の取り込み、および新たな脂質合成に対して独特の要件を示します。 代謝恒常性と系統の安定性を維持するために CTLA4 に依存します。
Strategies for therapeutic targeting of Treg cells for tumor therapy include their depletion and functional reprogramming by modulating co-inhibitory receptor (e.g., CTLA4), cytokine (e.g., IL-2), and chemokine receptor (e.g., CCR4 and CCR8) signaling.
腫瘍治療のために Treg 細胞を標的とする戦略には、共抑制受容体 (CTLA4 など)、サイトカイン (IL-2 など)、ケモカイン受容体 (CCR4 や CCR8 など) のシグナル伝達を調節することによる Treg 細胞の枯渇と機能再プログラミングが含まれます。
However, immune-associated adverse events remain a challenge for Treg cell-targeted cancer immunotherapies, with future therapeutics likely benefiting from a better understanding of tumor context-specific functional programming.
しかし、免疫関連の有害事象は、Treg 細胞を標的としたがん免疫療法にとって依然として課題です。 将来の治療法は、腫瘍の状況に応じた機能プログラミングをより深く理解することで恩恵を受ける可能性があります。
Additional studies are also warranted to ascertain the functional contributions of other immunosuppressive populations (e.g., Tr1 cells, KIR+ CD8+ T cells, and regulatory B cells) to immune health and disease, as their interplay with Treg cells is likely essential for balancing pro-inflammatory and immunosuppressive responses to different insults.
他の免疫抑制集団(Tr1 細胞、KIR+ CD8+ T 細胞、制御性 B 細胞など)の免疫の健康と疾患への機能的寄与を確認するために、追加の研究も必要です。 Treg細胞との相互作用は、さまざまな傷害に対する炎症促進性反応と免疫抑制性反応のバランスを取るために不可欠である可能性が高いためです。
How does the immune system balance tissue immunity and homeostasis?
免疫系は組織免疫と恒常性のバランスをどのようにとっているのでしょうか?
In a successful acute immune response, the offending agent is cleared, inflammation resolved, and homeostasis re-established.
急性免疫反応が成功すると、原因物質が排除され、炎症が解消され、恒常性が再確立されます。
Importantly, a primary immune response leaves the previously infected tissue more capable of defense against a subsequent infection.
重要なのは、一次免疫応答により、以前に感染した組織がその後の感染に対してより防御できる状態になることです。
Resident memory lymphocytes enter non-lymphoid tissues (though some also populate lymphoid tissues), where they perceive and respond to environmental cues and are maintained for long periods of time.
常駐する記憶リンパ球は非リンパ組織に入り(一部はリンパ組織にも存在しますが)、そこで環境の合図を感知して反応し、長期間維持されます。
They defend against future pathogen encounters and prevent the dangerous pathology associated with innate immune defenses triggered during a primary response.
これらは将来の病原体との遭遇を防御し、一次反応中に引き起こされる自然免疫防御に関連する危険な病態を防ぎます。
An inability to return to normal homeostatic set points results in chronic inflammation and multiple diseases, including allergic disease, fibrosis, autoimmunity, and neurodegenerative disorders, and such effects are counteracted by various intratissue and inter-tissue interactions between lymphocytes and tissue cells and factors.
正常な恒常性の設定値に戻れないと、慢性炎症や、アレルギー疾患、線維症、自己免疫、神経変性疾患などの複数の疾患が引き起こされます。 そして、そのような影響は、リンパ球と組織細胞および因子の間のさまざまな組織内および組織間の相互作用によって打ち消されます。
In this section, we discuss the diverse types of tissue lymphocytes and define how these “sentinels” contribute to defense against infection while supporting tissue function and organismal homeostasis.
このセクションでは、多様な種類の組織リンパ球について説明し、これらの「センチネル」が組織機能と生体恒常性をサポートしながら、感染に対する防御にどのように貢献するかを定義します。
Memory tissue lymphocytes are lifelong sentinels
記憶組織リンパ球は生涯の番兵である
Antigen-experienced lymphocytes form both circulating and TRM populations.
抗原を経験したリンパ球は、循環集団と TRM 集団の両方を形成します。
Antigen-specific TRM cells alter the rules of engagement for subsequent immune responses to the same invading agent, as these highly specialized cells clear pathogens at an exponentially faster rate than most innate mechanisms.
抗原特異的 TRM 細胞は、同じ侵入因子に対する後続の免疫応答の関与規則を変更します。これは、これらの高度に特殊化された細胞が、ほとんどの生来のメカニズムよりも指数関数的に速い速度で病原体を除去するためです。
Within hours of re-infection, TRM cells recruit, localize, and activate innate immune cells in tissues, a process that takes up to a day in their absence.
再感染から数時間以内に、TRM 細胞は組織内の自然免疫細胞を動員し、局在させ、活性化します。このプロセスには、自然免疫細胞が存在しない場合には最大 1 日かかります。
TRM and tissue-resident B (BRM) cells are anatomically, transcriptionally, epigenetically, and functionally distinct from their circulating counterparts.
TRM 細胞および組織常在 B (BRM) 細胞は、循環している対応する細胞とは解剖学的、転写的、エピジェネティックかつ機能的に異なります。
Although CD8+ TRM cells are well studied, there is also a growing interest in the more recently described CD4+ TRM and BRM cells.
CD8+ TRM 細胞はよく研究されていますが、より最近報告された CD4+ TRM 細胞および BRM 細胞にも関心が高まっています。
Resident memory cells are optimized to clear offending agents through their interactions with both the previously encountered offending agent and the tissue environment in which they need to functionally engage.
常駐記憶細胞は、以前に遭遇した攻撃因子と、それらが機能的に関与する必要がある組織環境の両方との相互作用を通じて、攻撃因子を排除するように最適化されています。
A two-site continuum of lymphocyte spatiotemporal differentiation occurs as the immune response progresses: lymphocytes are primed to gain functional properties in the lymphoid tissues and then learn how to communicate with their neighbors and serve as sentinels upon tissue entry.
免疫応答が進行するにつれて、リンパ球の 2 部位の時空間分化の連続が起こります:リンパ球は、リンパ組織内で機能的特性を獲得するように準備され、その後、隣接する細胞とコミュニケーションする方法を学習し、組織侵入時に番兵として機能します。
In both sites, differentiation processes occur such that broad classes of lymphocytes contribute to the memory pool: some that are more terminally differentiated with capacity to exert strong effector functions and others that serve as a more plastic, proliferative reserve population that can repopulate the effector-like pool.
どちらの部位でも、広範なクラスのリンパ球が記憶プールに寄与するように分化プロセスが発生します:強力なエフェクター機能を発揮する能力を備えてより最終的に分化したものもあれば、エフェクター様のプールを再構築できる、より可塑性の高い増殖予備集団として機能するものもあります。
Below, we discuss characteristics of lymphocytes that populate the tissues to provide protection against pathogens while also regulating homeostasis and tissue repair (Figure 4).
以下では、恒常性と組織修復を制御しながら、病原体に対する防御を提供するために組織に存在するリンパ球の特徴について説明します (図 4)。
Functional diversity of resident memory lymphocytes
常在記憶リンパ球の機能的多様性
CD8+ TRM cells are critical for imparting local immune protection against different infections.
CD8+ TRM 細胞は、さまざまな感染症に対する局所免疫防御を与えるために重要です。
Moreover, murine CD8+ TRM cells accelerate immune control of viral, bacterial, fungal, and parasitic infections.
さらに、マウス CD8+ TRM 細胞は、ウイルス、細菌、真菌、寄生虫感染症の免疫制御を促進します。
In humans, TRM cells are present in virtually every tissue examined.
ヒトでは、TRM 細胞は検査した事実上すべての組織に存在します。
Transcriptional profiling of human T cells from various tissue sites revealed a conserved transcriptional program that is distinct from blood memory T cells but shares key gene expression profiles with mouse TRM cells.
さまざまな組織部位からのヒトT細胞の転写プロファイリングにより、保存された転写プログラムが明らかになりました。これは血液メモリー T 細胞とは異なりますが、重要な遺伝子発現プロファイルはマウス TRM 細胞と共通しています。
In humans, the frequency of antigen-specific TRM cells correlates with better control of viruses, including hepatitis B virus in the liver, HIV in the lymphoid tissue, and RSV in the lung.
ヒトでは、抗原特異的 TRM 細胞の頻度は、肝臓の B 型肝炎ウイルス、リンパ組織の HIV、肺の RSV などのウイルスの制御の向上と相関しています。
Although most CD8+ TRM cells share some core signatures associated with migration and adhesion, there is also significant heterogeneity associated with the type of immunological insult that primes the response and the subsequent site of TRM residency.
ほとんどの CD8+ TRM 細胞は遊走と接着に関連するいくつかの核となる特徴を共有していますが、応答を開始する免疫学的損傷の種類とその後の TRM 常在部位に関連する重大な不均一性も存在します。
Because of this heterogeneity, diverse transcriptional and metabolic programs regulate the formation of each unique TRM population.
この不均一性のため、多様な転写および代謝プログラムがそれぞれの固有の TRM 集団の形成を制御します。
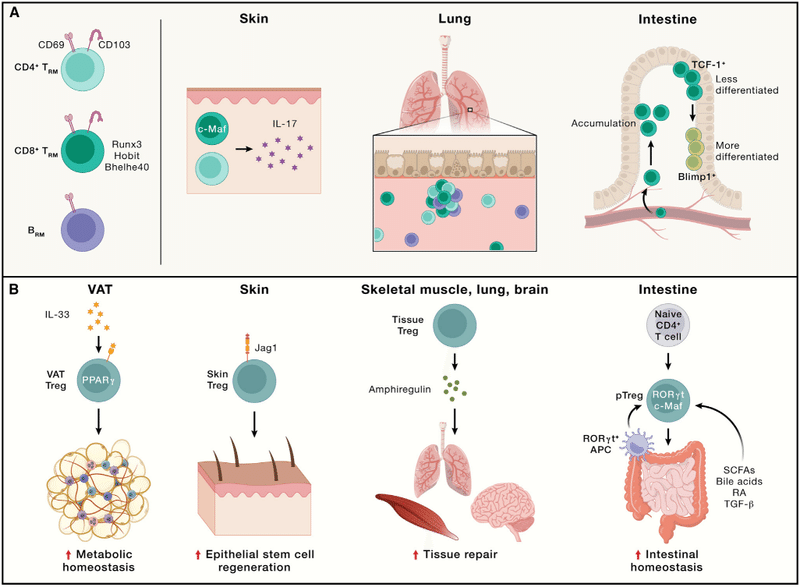
For example, deletion of the transcription factor Runx3 has varying effects on IFNγ-expressing CD8+ TRM numbers in distinct non-lymphoid tissues, and skin-resident CD8+ TRM cells that produce IL-17 upon bacterial infection and wound healing require c-Maf but not Runx3 for their generation.
たとえば、転写因子 Runx3 の欠失は、異なる非リンパ組織における IFNγ 発現 CD8+ TRM 数にさまざまな影響を及ぼします。 また、細菌感染や創傷治癒の際に IL-17 を産生する皮膚常駐 CD8+ TRM 細胞は、その生成に c-Maf を必要としますが、Runx3 は必要としません。
Intratissue heterogeneity even exists among antigen-specific TRM cells.
組織内不均一性は、抗原特異的 TRM 細胞間にも存在します。
For instance, CD8+ TRM cells within a tissue express varying levels of key TRM-associated molecules (e.g., CD69, CD103, IL18R, TCF-1, and T-bet), and the frequency of cells expressing these molecules can shift over time.
例えば、組織内の CD8+ TRM 細胞は、さまざまなレベルの重要な TRM 関連分子 (CD69、CD103、IL18R、TCF-1、T-bet など) を発現しており、これらの分子を発現する細胞の頻度は時間の経過とともに変化する可能性があります。
In the small intestine, two distinct subsets of TRM cells are observed: one with enhanced memory potential that expresses high levels of Id3 and TCF-1 but low levels of Blimp1, while the other expresses high levels of Blimp1 but low levels of Id3.
小腸では、TRM 細胞の 2 つの異なるサブセットが観察されます:1 つは、高レベルの Id3 と TCF-1 を発現するが、低レベルの Blimp1 を発現する強化された記憶能力を持ち、もう 1 つは、高レベルの Blimp1 を発現するが、低レベルの Id3 を発現します。
Of interest, CD8+ tumor-infiltrating lymphocytes (TILs) can also display characteristics of TRM cells in certain mouse and human cancers, which usually correlate with better antitumor immunity.
興味深いことに、CD8+ 腫瘍浸潤リンパ球 (TIL) は、特定のマウスおよびヒトのがんにおいて TRM 細胞の特徴を示すこともあります。 これは通常、抗腫瘍免疫の向上と相関します。
Understanding the distinctions between these subsets and how to properly regulate their distribution and function may therefore be pertinent to enhancing protective immunity against both infection and tumors.
したがって、これらのサブセット間の違いを理解し、それらの分布と機能を適切に調節する方法を理解することは、感染と腫瘍の両方に対する防御免疫を強化することに関連している可能性があります。
Although CD8+ TRM cells may be optimally poised to kill pathogens at the site of infection, CD4+ TRM cells are the great communicators of the system.
CD8+ TRM 細胞は感染部位で病原体を殺すために最適な状態にある可能性がありますが、CD4+ TRM 細胞はシステムの優れた伝達者です。
Like CD4+ T cells in a primary immune response, CD4+ TRM cells interact with many cell types through ligand-receptor interactions and secrete a diverse array of cytokines.
一次免疫応答における CD4+ T 細胞と同様に、CD4+ TRM 細胞はリガンドと受容体の相互作用を通じて多くの細胞型と相互作用し、多様なサイトカインを分泌します。
Moreover, functionally diverse CD4+ TRM populations contribute to the control of infections with viruses, bacteria, and parasites.
さらに、機能的に多様な CD4+ TRM 集団は、ウイルス、細菌、寄生虫による感染の制御に貢献します。
CD4+ TRM cells share transcriptional programs with CD8+ TRM cells and depend upon IL-2 signaling in the lymphoid tissue to become tissue resident.
CD4+ TRM 細胞は CD8+ TRM 細胞と転写プログラムを共有しており、リンパ組織内の IL-2 シグナル伝達に依存して組織常在化します。
CD4+ TRM cells can also undergo further diversification within the tissues, with some retaining a more effector-like phenotype and others behaving more like Tfh cells.
CD4+ TRM 細胞は組織内でさらに多様化することもあり、よりエフェクター様の表現型を保持する細胞もあれば、より Tfh 細胞に近い挙動を示す細胞もいます。
Whether this division of labor is a characteristic of only T-bet-expressing Th1-like TRM cells during influenza infection or is also a general feature of CD4+ TRM cells responding to other types of immunologic insults requires further investigation.
この分業がインフルエンザ感染時のT-bet発現Th1様TRM細胞のみの特徴であるのか、それとも他の種類の免疫学的傷害に応答するCD4+ TRM細胞の一般的な特徴でもあるのかどうかについては、さらなる研究が必要である。
BRM cells can also protect against re-infection.
BRM 細胞は再感染を防ぐこともできます。
Recent studies using fluorescently labeled probes have characterized influenza-specific memory B cells and begun to shed light on the phenotype, longevity, functional attributes, and interactions with other tissue-resident immune populations.
蛍光標識プローブを使用した最近の研究では、インフルエンザ特異的メモリー B 細胞の特徴が解明され、表現型、寿命、機能的特性、および他の組織常在免疫集団との相互作用が解明され始めています。
BRM cells are randomly distributed throughout the lung, near local alveoli.
BRM 細胞は、肺全体の局所肺胞近くにランダムに分布しています。
Upon re-infection, these activated BRM cells rapidly migrate to sites of infection, where they produce antibodies to help clear the virus.
再感染すると、これらの活性化された BRM 細胞は感染部位に急速に移動し、そこでウイルスの除去を助ける抗体を産生します。
A survey of mouse and human tissues characterized Bmem cells in the lung and gut tissues that express both shared and unique phenotypic markers, which will aid our understanding of BRM cells.
マウスとヒトの組織を調査した結果、共通の表現型マーカーと独自の表現型マーカーの両方を発現する肺および腸組織の Bmem 細胞の特徴が明らかになりました。 これは BRM 細胞の理解を助けるでしょう。
Tissue Treg cells support tissue homeostasis and repair
組織Treg細胞は組織の恒常性と修復をサポートします
The optimal functionality of TRM cells can be a double-edged sword by providing immune protection and contributing to immunopathology.
TRM 細胞の最適な機能は、免疫保護を提供し、免疫病理学に寄与することにより、両刃の剣となり得ます。
For instance, CD4+ TRM cells can promote allergic and autoimmune diseases of the skin, lung, nervous system, and gut.
たとえば、CD4+ TRM 細胞は、皮膚、肺、神経系、腸のアレルギー疾患や自己免疫疾患を促進する可能性があります。
As such, Treg cells contribute to counteracting these effects by promoting tissue homeostasis, as well as regeneration and repair in response to various insults.
そのため、Treg 細胞は組織の恒常性を促進し、さまざまな傷害に応答して再生と修復を促進することで、これらの影響に対抗することに貢献します。
Treg cells in non-lymphoid tissues, such as those in the visceral adipose tissue (VAT), adapt to specific microenvironmental signals, and accordingly, VAT Treg cells have distinct transcriptome profiles (partly mediated by PPARγ and IL-33) and TCR repertoires than splenic Treg cells.
内臓脂肪組織 (VAT) などの非リンパ組織の Treg 細胞は、特定の微小環境シグナルに適応します。 したがって、VAT Treg 細胞は、脾臓 Treg 細胞とは異なるトランスクリプトーム プロファイル (部分的に PPARγ および IL-33 によって媒介される) および TCR レパートリーを持っています。
These VAT Treg cells orchestrate tissue homeostasis via local and systemic regulation of metabolism, partly by downmodulating tissue inflammation that may predispose to type 2 diabetes and metabolic syndromes, and their generation requires a stepwise, multi-site differentiation process.
これらの VAT Treg 細胞は、部分的には 2 型糖尿病やメタボリックシンドロームの素因となる組織の炎症を下方調節することによって、局所的および全身的な代謝調節を介して組織の恒常性を調整します。 そしてそれらの生成には、段階的な複数サイトの分化プロセスが必要です。
Beyond VAT, scRNA-seq profiling reveals tissue-specific signatures of Treg cells and their progressive adaptation to tissue sites under steady state.
VAT を超えて、scRNA-seq プロファイリングにより、Treg 細胞の組織特異的な特徴と定常状態での組織部位へのそれらの漸進的な適応が明らかになります。
Tissue Treg cells also promote tissue regeneration via interfacing with stem cells.
組織 Treg 細胞は、幹細胞とのインターフェースを介して組織再生も促進します。
For example, in the skin, Treg cells localize to hair follicles, where they stimulate stem cell function or regeneration via Notch signaling.
たとえば、皮膚では、Treg 細胞は毛包に局在し、そこで Notch シグナル伝達を介して幹細胞の機能や再生を刺激します。
In a mouse model of vitiligo, the proper position and function of skin Treg cells are mediated by CCR5 signaling, which likely extends to human vitiligo patients.
白斑のマウスモデルでは、皮膚 Treg 細胞の適切な位置と機能は CCR5 シグナル伝達によって媒介されており、これはヒトの白斑患者にも当てはまると考えられます。
Finally, in response to tissue-specific insults, Treg cells support tissue repair and physiological function by production of growth factors.
最後に、組織特異的な損傷に応答して、Treg 細胞は成長因子の産生によって組織修復と生理学的機能をサポートします。
In particular, upon tissue injury, Treg cells produce amphiregulin to promote tissue repair and regeneration in the skeletal muscle, lung, and brain.
特に、組織が損傷すると、Treg 細胞はアンフィレグリンを生成して、骨格筋、肺、脳の組織修復と再生を促進します。
In a mouse model of influenza infection, Treg cell-derived amphiregulin prevents lung tissue damage without affecting antiviral immune responses, revealing discrete functions of Treg cells in mediating immune suppression and tissue repair.
インフルエンザ感染のマウスモデルでは、Treg細胞由来のアンフィレグリンが抗ウイルス免疫反応に影響を与えることなく肺組織の損傷を防ぎます。 免疫抑制と組織修復の仲介における Treg 細胞の個別の機能が明らかになりました。
The extent to which Treg cells interplay with tissue cells and impact physiological function and tissue repair, and the underlying processes, are fruitful areas of future studies.
Treg 細胞がどの程度組織細胞と相互作用し、生理学的機能や組織修復、およびその根底にあるプロセスに影響を与えるかは、今後の研究にとって有益な分野です。
Commensal microbes interplay with the immune system in barrier tissues, and specialized tissue Treg cells called peripheral Treg (pTreg) cells prevent immune reactivity to microbiota to preserve tissue homeostasis.
共生微生物はバリア組織の免疫系と相互作用しており、末梢 Treg (pTreg) 細胞と呼ばれる特殊な組織 Treg 細胞が微生物叢に対する免疫反応を阻止して組織の恒常性を維持します。
Upon antigen stimulation, pTreg cells are derived from naive T cells in response to commensal microbe-derived signals (e.g., short-chain fatty acids, secondary bile acids, and retinoic acid) or TGF-β.
抗原刺激により、pTreg 細胞はナイーブ T 細胞から派生します。 共生微生物由来のシグナル (短鎖脂肪酸、二次胆汁酸、レチノイン酸など) または TGF-β に応答してます。
In the intestines, these pTreg cells are marked by RORγt and c-Maf expression.
腸では、これらの pTreg 細胞は RORγt および c-Maf の発現によって特徴付けられます。
Exposure to food antigens also induces pTreg cell differentiation.
食物抗原への曝露も pTreg 細胞の分化を誘導します。
RORγt+ APCs are crucial for intestinal pTreg cell generation, in part, via integrin αVβ8 expression that enables TGF-β activation.
RORγt+ APC は腸内 pTreg 細胞の生成に重要です。 部分的には、TGF-βの活性化を可能にするインテグリンαVβ8の発現を介します。
The identification of the interactions between RORγt+ APCs and pTreg cells reveals an important mechanism for peripheral tolerance that distinguishes commensal microbes from invading harmful insults.
RORγt+ APC と pTreg 細胞の間の相互作用の同定により、共生微生物を有害な傷害の侵入から区別する末梢寛容の重要な機構が明らかになりました。
The prevalence and functionality of pTreg cells and their corresponding APCs in other tissues warrant further investigation.
他の組織における pTreg 細胞およびそれらに対応する APC の普及率と機能については、さらなる研究が必要です。
Where do lymphocytes fit into organismal homeostasis?
リンパ球は生体の恒常性のどこに当てはまるのでしょうか?
Organismal homeostasis maintains the internal stability of the host while adjusting for changing external conditions, including alterations in temperature, exposure to toxins or infectious agents, or dietary changes.
生物の恒常性は、温度の変化、毒素や感染因子への曝露、食事の変更などの外部条件の変化に合わせて調整しながら、宿主の内部安定性を維持します。
Sensing of these external insults by the immune system not only activates direct defense mechanisms, but if life is perceived to be threatened, also engages both local and system-wide changes to clear the offending agent.
免疫系によるこれらの外部攻撃の感知は、直接的な防御機構を活性化するだけでなく、生命が脅かされていると認識されると、局所的およびシステム全体の両方の変化を引き起こして、攻撃因子を排除します。
In a physiological emergency (e.g., infection with a replicating pathogen and injury or tissue stress), the immune system overrides the normal set points of homeostasis to help control the infection or heal the injury, a process referred to as inflammation.
生理学的緊急事態(例、複製する病原体による感染および損傷または組織ストレス)では、免疫系が正常な恒常性の設定値を無効にして、感染の制御または損傷の治癒を助けます。これは炎症と呼ばれるプロセスです。
The immune system also contributes to the re-establishment of organismal homeostasis through interactions with the other systems of the body.
免疫システムは、体の他のシステムとの相互作用を通じて生物の恒常性の再確立にも貢献します。
As discussed below, communication between the immune and nervous systems creates a complex homeostatic network that synergistically enhances the individual function of each system.
以下で説明するように、免疫系と神経系の間のコミュニケーションは、各システムの個々の機能を相乗的に強化する複雑な恒常性ネットワークを作成します。
Although innate lymphoid cells (ILCs) contribute to sensing and responding to perturbations to homeostasis in an antigen-independent manner, recent studies have identified similar types of interactions between lymphocytes and the nervous system that depend upon sensing of a specific antigen, with major implications for therapeutic interventions.
自然リンパ球 (ILC) は、抗原非依存的な方法で恒常性の乱れを感知して応答することに貢献していますが、最近の研究では、特定の抗原の感知に依存するリンパ球と神経系との間の同様の種類の相互作用が特定されています。 治療介入に大きな影響を及ぼします。
Lymphocytes and neurons communicate using a common language of cytokines, hormones, neuropeptides, chemokines, and their receptors, and neuroimmune interactions are observed in the central and peripheral nervous systems.
リンパ球とニューロンは、サイトカイン、ホルモン、神経ペプチド、ケモカイン、およびそれらの受容体という共通言語を使用して通信し、中枢神経系と末梢神経系では神経免疫相互作用が観察されます。
Recent evidence has highlighted the ability of antigen-specific lymphocytes to direct neuronal activity in multiple tissue sites.
最近の証拠により、抗原特異的リンパ球が複数の組織部位でニューロン活動を指示する能力が強調されています。
In the skin dermis, IL-17-producing CD4+ TRM cells that are specific for commensal microbiota co-localize with sensory nerve fibers, where they promote axonal growth and nerve regeneration upon injury.
皮膚の真皮では、共生微生物叢に特異的な IL-17 産生 CD4+ TRM 細胞が感覚神経線維と共局在しています。 損傷時に軸索の成長と神経の再生を促進します。
In the intestine, sensitization to food allergens drives antigen-specific avoidance behavior that depends upon B cell-secreted IgE, suggesting that antibodies may expand the sensory capacity of the nervous system.
腸では、食物アレルゲンに対する感作により、B 細胞から分泌される IgE に依存する抗原特異的な回避行動が引き起こされます。 これは、抗体が神経系の感覚能力を拡張する可能性があることを示唆しています。
Remarkably, this allergen-specific response persists for at least 48 weeks after allergic sensitization, further suggesting a specific role for gut-resident memory lymphocytes.
驚くべきことに、このアレルゲン特異的反応は、アレルギー感作後少なくとも 48 週間持続します。 さらに、腸内に存在する記憶リンパ球の特定の役割が示唆されています。
However, these additive responses are not always protective.
ただし、これらの追加反応は常に保護的であるとは限りません。
For example, IL-5Rα-expressing nociceptive nerves in the lung are activated by IL-5 produced by ILC2s and Th2 cells, creating a feedforward loop that enhances immunopathology.
たとえば、肺の IL-5Rα を発現する侵害受容神経は、ILC2 および Th2 細胞によって産生される IL-5 によって活性化され、免疫病理を増強するフィードフォワード ループを作り出します。
The peripheral nervous system also modulates immune responses in lymphoid and non-lymphoid organs.
末梢神経系は、リンパ系および非リンパ系臓器の免疫応答も調節します。
In response to fasting, catecholaminergic neurons are activated in the ventrolateral medulla, which can shunt CD4+ and CD8+ T cells from the blood and secondary lymphoid organs into the bone marrow in a CXCR4-dependent manner.
絶食に反応して、延髄腹側外側のカテコールアミン作動性ニューロンが活性化されます。 これは、CXCR4依存的にCD4+およびCD8+ T細胞を血液および二次リンパ器官から骨髄に分流させることができます。
Catecholamines contribute to CD8+ T cell exhaustion in both chronic viral infection and cancer, further providing evidence of the intersection of neuronal regulation of the tissue immune response.
カテコールアミンは、慢性ウイルス感染症とがんの両方における CD8+ T 細胞の枯渇に寄与し、組織免疫応答のニューロン制御の交差の証拠をさらに提供します。
Specifically, exhausted antigen-specific CD8+ T cells are in close proximity to sympathetic nerves, and ablation of β-adrenergic signaling synergizes with ICB to improve T cell effector function, revealing a potential new therapeutic modality.
具体的には、抗原特異的 枯渇CD8+ T 細胞は交感神経に近接しており、β アドレナリン作動性シグナル伝達の除去が ICB と相乗して T 細胞エフェクター機能を改善します。 潜在的な新しい治療法を明らかにします。
Together, these studies highlight the importance of understanding neuroimmune interactions in tissues and in the larger context of how the body responds to homeostatic perturbations across all systems.
これらの研究を総合すると、組織における神経免疫相互作用と、すべてのシステムにわたる恒常性の乱れに対して身体がどのように反応するかというより大きな文脈において、神経免疫相互作用を理解することの重要性が強調されています。
How are distinct immune responses regulated?
異なる免疫反応はどのように制御されるのでしょうか?
Adaptive immune cells employ similar and unique strategies against different threats, orchestrated by extrinsic and intrinsic molecular processes that vary across space and time (Figure 5).
適応免疫細胞は、時空を超えて変化する外因性および内因性の分子プロセスによって調整された、さまざまな脅威に対して同様の独自の戦略を採用します (図 5)。
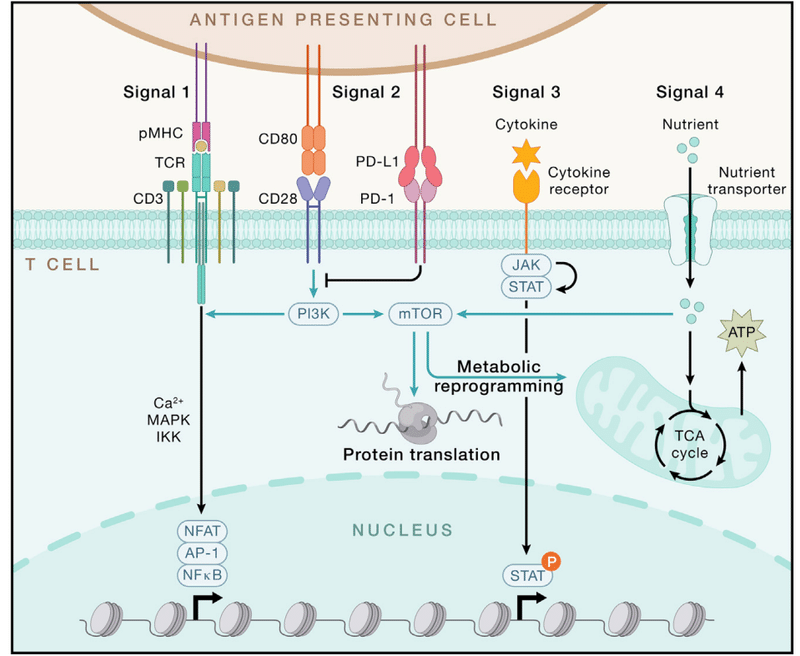
The extrinsic signals from antigens, co-stimulation, and cytokines (signals 1–3) are predominantly delivered from activated DCs.
抗原、共刺激、およびサイトカインからの外因性シグナル (シグナル 1 ~ 3) は、主に活性化された DC から伝達されます。
Nutrients (signal 4) and other cellular interactions also regulate lymphocyte specialization.
栄養素 (シグナル 4) およびその他の細胞相互作用もリンパ球の特殊化を制御します。
These extrinsic cues are integrated by cell-intrinsic signaling pathways, metabolism, and epigenetic programs, which culminate in altered gene transcription and protein translation instructing cell state and fate.
これらの外因性の合図は、細胞固有のシグナル伝達経路、代謝、およびエピジェネティックなプログラムによって統合され、最終的に細胞の状態と運命を指示する遺伝子転写とタンパク質翻訳の変化をもたらします。
Extrinsic signals drive adaptive immunity
外部シグナルが適応免疫を促進する
The diverse stimuli of adaptive immunity largely converge on signals 1–3, which mediate the specificity, strength, and durability of immune responses (Figure 5).
適応免疫の多様な刺激は主にシグナル 1 ~ 3 に集中しており、これらのシグナルが免疫応答の特異性、強度、持続性を媒介します (図 5)。
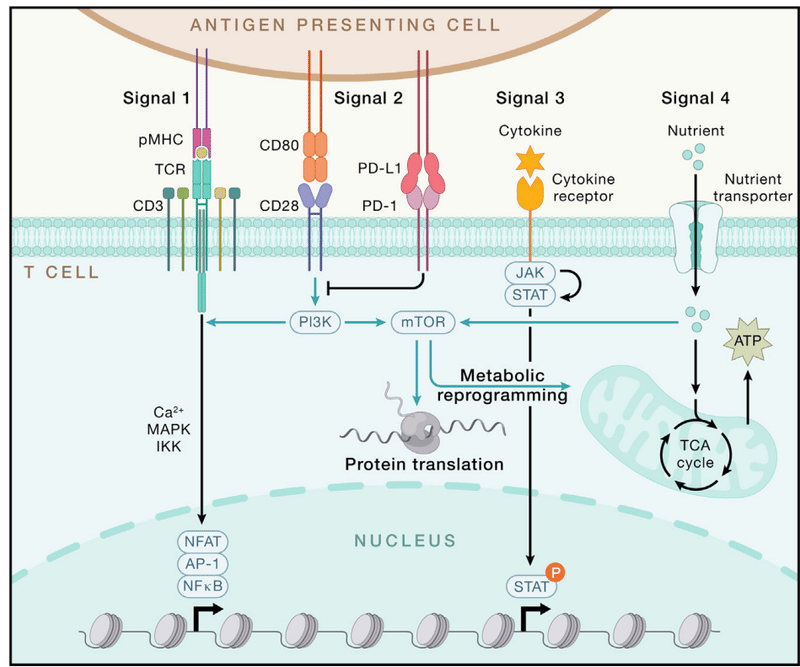
During naive T cell activation, TCR stimulation (signal 1) occurs upon recognition of DC-presented pMHC, leading to immunological synapse formation, which shapes TCR signaling and mechanical force.
ナイーブ T 細胞の活性化中に、DC によって提示された pMHC の認識により TCR 刺激 (シグナル 1) が発生し、TCR シグナル伝達と機械的力を形成する免疫学的シナプス形成を引き起こします。
Downstream of antigen receptors, graded IRF4 expression establishes the fate and function of CD8+ T cells, CD4+ T cells, and B cells.
抗原受容体の下流では、段階的な IRF4 発現によって、CD8+ T 細胞、CD4+ T 細胞、および B 細胞の運命と機能が確立されます。
Acute and persistent antigen stimulation promote the generation of effector and exhausted T cells, respectively.
急性および持続的な抗原刺激は、それぞれエフェクター細胞および枯渇 T 細胞の生成を促進します。
Of note, pMHC-TCR interaction is among the most complex ligand-receptor interaction systems for directing specific biological responses, associated with the vast diversity of antigens and antigen receptors as described above.
注目すべきことに、pMHC-TCR相互作用は、特定の生物学的応答を導くための最も複雑なリガンド-受容体相互作用システムの1つであり、上記のような非常に多様な抗原および抗原受容体に関連しています。
However, TCR signaling is not sufficient to elicit a productive adaptive immune response, and this serves to prevent spurious T cell activation and uncontrolled inflammation.
ただし、TCR シグナル伝達は生産的な適応免疫応答を誘発するには十分ではありません。 これは、偽の T 細胞活性化や制御不能な炎症を防ぐ働きをします。
Instead, co-stimulatory (signal 2) and cytokine (signal 3) signals act in coordination with antigen stimulation to instruct adaptive immunity.
代わりに、共刺激 (シグナル 2) およびサイトカイン (シグナル 3) シグナルが抗原刺激と協調して作用し、適応免疫を指示します。
For example, CD28 (signal 2) and IL-12 (signal 3) help promote naive T cell activation.
たとえば、CD28 (シグナル 2) と IL-12 (シグナル 3) は、ナイーブ T 細胞の活性化の促進に役立ちます。
Other co-stimulatory (e.g., ICOS) and co-inhibitory signals (e.g., PD-1, CTLA4, LAG3, TIM-3, and TIGIT) also serve as respective “accelerators” and “brakes” for signal 2.
他の共刺激シグナル (ICOS など) および共抑制シグナル (PD-1、CTLA4、LAG3、TIM-3、TIGIT など) も、シグナル 2 のそれぞれの「アクセル」および「ブレーキ」として機能します。
For signal 3, immunostimulatory (e.g., IL-1, IL-2, and IL-6) and immunosuppressive (e.g., TGF-β and IL-10) cytokines shape T cell fate.
シグナル 3 については、免疫刺激性サイトカイン (IL-1、IL-2、IL-6 など) および免疫抑制性サイトカイン (TGF-β および IL-10 など) が T 細胞の運命を決定します。
Importantly, the interplay between signals 1–3 orchestrates adaptive immune responses, including by activating metabolic rewiring (e.g., via TCR and CD28) or tuning downstream signaling (e.g., ICOS-mediated phosphatidylinositol 3-kinase [PI3K] activation for Tfh responses).
重要なことに、シグナル 1 ~ 3 間の相互作用は、代謝再配線の活性化 (例: TCR および CD28 経由) や下流シグナル伝達の調整 (例: Tfh 応答に対する ICOS 媒介ホスファチジルイノシトール 3-キナーゼ [PI3K] 活性化) などにより、適応免疫応答を調整します。
Also, PD-1-targeted ICB therapies enhance CD28-dependent effector function and accumulation of CD8+ T cells in chronic infection and cancer and also act in synergy with IL-2 signaling.
また、PD-1 を標的とした ICB 療法は、慢性感染症やがんにおける CD28 依存性エフェクター機能と CD8+ T 細胞の蓄積を強化し、IL-2 シグナル伝達と相乗的に作用します。
How these co-stimulatory and cytokine signals are delivered by specific innate stimuli and cells, including “non-traditional” APCs (e.g., epithelial cells), as well as the impact of other non-traditional signals (e.g., serum amyloid A proteins derived from intestinal epithelial cells that act in collaboration with Th17 cell-promoting cytokines), will be important to explore.
これらの共刺激シグナルとサイトカインシグナルが、「非伝統的な」APC(例:上皮細胞)を含む特定の先天性刺激や細胞によってどのように伝達されるか、また他の非伝統的なシグナル(例:Th17細胞促進サイトカインと連携して作用する腸上皮細胞由来の血清アミロイドAタンパク質)の影響も調査することが重要です。
Nutrients and metabolites license T cell immunity by functioning as signal 4, including acting on intratumoral CD8+ T cells.
栄養素と代謝物はシグナル 4 として機能することで T 細胞免疫を活性化します。 腫瘍内 CD8+ T 細胞への作用を含みます。
In particular, glucose and glutamine are limiting for intratumoral lymphocytes due to the enhanced capacities of intratumoral myeloid cells and tumors cells to acquire and consume these nutrients.
特に、グルコースとグルタミンは腫瘍内リンパ球を制限します。 これは、腫瘍内骨髄細胞と腫瘍細胞がこれらの栄養素を獲得して消費する能力が強化されているためです。
Consequently, intratumoral glucose restriction impairs T cell activation and function.
その結果、腫瘍内グルコース制限は T 細胞の活性化と機能を損ないます。
Similarly, glutamine shapes metabolic reprogramming and context-specific function of T cells, as well as intratumoral cDC1s.
同様に、グルタミンは、T 細胞および腫瘍内 cDC1 の代謝再プログラミングと状況特異的機能を形成します。
Intratumoral glutamine administration rectifies defective cDC1-dependent CD8+ T cell antitumor immunity and overcomes therapeutic resistance to immunotherapies in animal models.
腫瘍内グルタミン投与は、動物モデルにおける欠陥のあるcDC1依存性CD8+ T細胞抗腫瘍免疫を修正し、免疫療法に対する治療抵抗性を克服します。
Other amino acids, such as arginine and methionine, are also required for T cell function, partly by orchestrating mTORC1 activation, whereas uptake of lactic acid and oxidized lipids is largely detrimental to intratumoral T cell function.
アルギニンやメチオニンなどの他のアミノ酸も、部分的にmTORC1活性化を調整することによってT細胞機能に必要ですが、乳酸や酸化脂質の取り込みは腫瘍内T細胞機能に大きな悪影響を及ぼします。
Finally, G-protein-coupled receptors orchestrate T cell immunity, partly by mediating signals from stress-associated hormones (e.g., catecholamines) or the dietary nutrient trans-vaccenic acid.
最後に、G タンパク質共役受容体は、ストレス関連ホルモン (カテコールアミンなど) または食事栄養素トランス バクセン酸からのシグナルを部分的に媒介することによって、T 細胞免疫を調整します。
Future research should explore how adaptive immunity is shaped by nutrient availability and usage in various tissues or from dietary and microbiota-derived sources.
今後の研究では、さまざまな組織における栄養素の利用可能性と利用、または食事や微生物叢由来の供給源からの栄養素の利用可能性と利用によって、適応免疫がどのように形成されるかを調査する必要があります。
Cell-intrinsic programs
細胞固有のプログラム
How do lymphocytes interpret and integrate these diverse signals?
リンパ球はこれらの多様なシグナルをどのように解釈し、統合するのでしょうか?
Antigen and co-stimulatory receptor engagement induces a series of phosphorylation events and other post-translational modifications, leading to the activation of key transcription factors nuclear factor κB (NF-κB) (via IKK signaling), AP-1 (via ERK signaling), and NFAT (via calcium-calcineurin signaling).
抗原と共刺激受容体の結合により、一連のリン酸化イベントやその他の翻訳後修飾が誘導されます。 主要な転写因子である核因子κB (NF-κB) (IKK シグナル伝達経由)、AP-1 (ERK シグナル伝達経由)、および NFAT (カルシウム カルシニューリン シグナル伝達経由) の活性化につながります。
Additionally, cytokines primarily activate the JAK-STAT pathway, while nutrient-dependent signaling is mediated by a three-tier process composed of nutrient transporters, sensors, and transducers.
さらに、サイトカインは主に JAK-STAT 経路を活性化しますが、栄養素依存性シグナル伝達は、栄養素輸送体、センサー、トランスデューサーからなる 3 層プロセスによって媒介されます。
As with signals 1–4 themselves, crosstalk and integration of such signaling pathways regulate adaptive immunity, as illustrated by the cooperation between NFAT and AP-1 or STAT and lineage-specific transcription factors, thereby leading to transcriptional activation of signature cytokines and effector molecules.
シグナル 1 ~ 4 自体と同様に、NFAT と AP-1 または STAT と系統特異的転写因子の間の連携によって示されるように、このようなシグナル伝達経路のクロストークと統合が適応免疫を制御します。 それにより、特徴的なサイトカインおよびエフェクター分子の転写活性化が引き起こされます。
Feedback control mechanisms also exist to prevent T cell hyperactivation, including those mediated by the Cbl family proteins and Regnase-1.
Cbl ファミリータンパク質や Regnase-1 によって媒介されるものなど、T 細胞の過剰活性化を防ぐためのフィードバック制御機構も存在します。
In particular, emerging studies highlight the important roles of metabolic and epigenetic rewiring that shape gene transcription and cell fate choices.
特に、新たな研究は、遺伝子転写と細胞運命の選択を形作る代謝的およびエピジェネティックな再配線の重要な役割を強調しています。
Metabolic regulation of adaptive immunity has been revealed over the past decade.
適応免疫の代謝調節は、過去 10 年間にわたって明らかにされてきました。
Glycolysis promotes effector over Treg cell differentiation, and fatty acid metabolism contributes to memory T cell generation.
解糖は Treg 細胞の分化よりもエフェクターを促進し、脂肪酸代謝はメモリー T 細胞の生成に寄与します。
Also, mitochondrial dysregulation is a hallmark of exhausted CD8+ T cells.
また、ミトコンドリアの調節不全は、枯渇CD8+ T 細胞の特徴です。
Mechanistically, extensive crosstalk between immune signaling and metabolic programs directs “bidirectional metabolic signaling.”
機構的には、免疫シグナル伝達と代謝プログラムの間の広範なクロストークが「双方向の代謝シグナル伝達」を導きます。
For instance, mTORC1 integrates signals 1–4 and shows reciprocal interplay with Myc, thereby driving anabolic metabolism, quiescence exit, and T cell differentiation.
たとえば、mTORC1 はシグナル 1 ~ 4 を統合し、Myc との相互作用を示し、それによって同化代謝、静止状態からの脱出、および T 細胞の分化を促進します。
Beyond generating ATP, mitochondria are signaling hubs in T cell biology, as specific mitochondrial metabolites (e.g., α-ketoglutarate and the oncometabolite d-2-hydroxyglutarate) impact the epigenetic landscape or protein activity in T cells.
特定のミトコンドリア代謝産物(α-ケトグルタル酸や腫瘍代謝産物のd-2-ヒドロキシグルタル酸など)がT細胞のエピジェネティックな状況やタンパク質活性に影響を与えるため、ミトコンドリアはATPの生成以外にもT細胞生物学におけるシグナル伝達のハブとなっています。
Moreover, new metabolic regulators of T cell fate, including phosphatidylethanolamine and guanosine diphosphate (GDP)-fucose signaling, which respectively drive Tfh and terminal effector CD8+ T cell differentiation, were identified by CRISPR-based genetic screens.
さらに、T 細胞運命の新しい代謝調節因子が CRISPR ベースの遺伝子スクリーニングによって同定されました。ホスファチジルエタノールアミンおよびグアノシン二リン酸 (GDP)-フコースシグナル伝達が含まれますが、 これらはそれぞれ、Tfh およびターミナルエフェクター CD8+ T 細胞の分化を促進するものであり、CRISPR ベースの遺伝子スクリーニングによって同定されました。
Altogether, cell state or fate is characterized by unique metabolic profiles, nutrient requirements, and tissue-specific regulators.
全体として、細胞の状態または運命は、固有の代謝プロファイル、栄養素要件、および組織固有の調節因子によって特徴付けられます。
Metabolic adaptation represents a key means to alter the fitness and function of adaptive immune cells to the specific immunological context.
代謝適応は、特定の免疫学的状況に対する適応免疫細胞の適合性と機能を変更するための重要な手段となります。
Epigenetic remodeling confers phenotypic stability of immune cells and is more reflective of cell fate alterations than transcriptional and metabolic events.
エピジェネティックなリモデリングは免疫細胞の表現型の安定性をもたらし、転写や代謝のイベントよりも細胞運命の変化をより反映します。
As such, epigenetic analyses can resolve key questions in adaptive immune responses.
したがって、エピジェネティック分析は、適応免疫応答における重要な問題を解決できます。
For instance, chromatin state analyses show that exhausted CD8+ T cells represent a separate T cell lineage in chronic infection and cancer.
たとえば、クロマチン状態分析では、枯渇 CD8+ T 細胞が慢性感染症とがんにおける別の T 細胞系統を表すことが示されています。
Chromatin accessibility analyses also revealed the progressive differentiation of exhausted T cells, while global mapping of histone methylation in effector CD4+ T cells exposes subset-specific patterns of signature cytokines and plasticity for master transcription factors.
クロマチンアクセシビリティ分析では、枯渇 T 細胞の分化が進行していることも明らかになりました。一方、エフェクター CD4+ T 細胞におけるヒストンメチル化の全体的なマッピングにより、特徴的なサイトカインのサブセット特異的なパターンとマスター転写因子の可塑性が明らかになりました。
Accordingly, targeting the DNA methyltransferase Dnmt3a alters CD8+ T cell differentiation in acute and chronic responses, while disruption of the chromatin remodeling complex SWI/SNF affects CD8+ T cell differentiation and Treg cell activation and function.
したがって、DNAメチルトランスフェラーゼDnmt3aを標的とすることは、急性および慢性反応におけるCD8+ T細胞の分化を変化させる一方、クロマチンリモデリング複合体SWI/SNFの破壊はCD8+ T細胞の分化とTreg細胞の活性化および機能に影響を与えます。
These findings offer new opportunities for therapeutic targeting of epigenetic programs for cancer and immune-mediated diseases.
これらの発見は、がんや免疫介在性疾患に対するエピジェネティックプログラムの治療標的化に新たな機会を提供します。
Engineering immunity: Vaccines and therapeutics
免疫工学: ワクチンと治療法
The hallmarks of T cell and B cell responses (cytotoxic and helper functions and antibody secretion) are the core components mediating effective and durable protection in the context of vaccines.
T 細胞と B 細胞の応答 (細胞傷害性機能とヘルパー機能、抗体分泌) の特徴は、ワクチンの効果的かつ持続的な防御を媒介する中心的な要素です。
Further, these hallmarks are leveraged as tools for therapeutic interventions, including the use of T cell-based adoptive cell therapies (ACTs) as “living drugs” and monoclonal antibodies (mAbs).
さらに、これらの特徴は、「生きた薬」としての T 細胞ベースの養子細胞療法 (ACT) やモノクローナル抗体 (mAb) の使用など、治療介入のツールとして活用されています。
Harnessing the power and unique features of the adaptive immune system holds great promise to treat a broad range of diseases, which is briefly described below mainly from the perspectives of vaccination for infectious diseases and engineered therapies for cancer.
適応免疫系の力と独特の特徴を利用することは、広範囲の疾患の治療に大きな期待を寄せています。これについては、主に感染症のワクチン接種とがんの工学的治療の観点から以下で簡単に説明します。
Vaccination as a means to understand and utilize adaptive immunity
適応免疫を理解し活用する手段としてのワクチン接種
Vaccination is, in many ways, where the understanding of adaptive immunity began, with the Jennerian cowpox vaccine for smallpox.
ワクチン接種は、さまざまな意味で、適応免疫の理解が始まった場所です。 天然痘用のジェンネリアン牛痘ワクチンが使用されました。
Vaccines aim to generate protective immune memory that will aid clearance and limit the pathology and inflammation of an infectious challenge.
ワクチンは、感染症の除去を助け、病状や炎症を制限する防御免疫記憶を生成することを目的としています。
The majority of vaccines are benchmarked on their ability to induce serum antibody responses, which are generally strong correlates of protection for most vaccines.
大部分のワクチンは、血清抗体反応を誘導する能力に基づいてベンチマークされており、これは一般に、ほとんどのワクチンの防御と強い相関関係があります。
While animal models and human studies have reported correlations between vaccine efficacy and T cell response features, including magnitude and quality in various systems, there are no rigorously validated, epidemiologically established T cell-based correlates of protection for any vaccine in clinical use.
動物モデルとヒトの研究では、ワクチンの有効性と、さまざまなシステムにおける大きさや質を含むT細胞応答特性との相関関係が報告されていますが、 臨床使用されているワクチンについては、厳密に検証され、疫学的に確立された T 細胞ベースの防御相関は存在しません。
This is likely because assays for T cell enumeration and characterization are more time consuming, expensive, and difficult to compare than antibody assays.
これは、T 細胞の計数と特性評価のアッセイが抗体アッセイよりも時間がかかり、高価であり、比較するのが難しいためであると考えられます。
Although few studies have directly compared the predictive power of antibodies versus cellular responses in human patients, the contributions of CD4+ and CD8+ T cells versus B cells can be defined mechanistically with genetic ablation and depletion studies in animal models.
抗体の予測力とヒト患者の細胞反応を直接比較した研究はほとんどありませんが、CD4+ および CD8+ T 細胞と B 細胞の寄与は、動物モデルでの遺伝子除去および枯渇研究により機構的に定義できます。
The recent interest of defining correlates of protection to COVID-19 vaccines has highlighted the utility of animal models for determining the precise mechanisms of protection and the need for improved human assays that can assess multiple arms of immunity.
COVID-19ワクチンに対する防御の相関関係を定義するという最近の関心は、防御の正確なメカニズムを決定するための動物モデルの有用性と、複数の免疫部門を評価できる改良されたヒトアッセイの必要性を浮き彫りにしている。
The understanding of human vaccinology has advanced significantly during the COVID-19 pandemic.
COVID-19パンデミックの中で、ヒトのワクチン学の理解は大幅に進歩しました。
One striking finding concerned the length of germinal center reactions after mRNA vaccination against COVID-19 that has revolutionized modern vaccine development.
顕著な発見の 1 つは、現代のワクチン開発に革命をもたらした、COVID-19 に対する mRNA ワクチン接種後の胚中心反応の長さに関するものでした。
In mouse studies, most germinal center reactions induced by immunization are resolved within 6 weeks; however, in humans, naive COVID-19 mRNA vaccine responses for both Tfh and B cells are still detected at 6 months after boosting, when the response is peaking in many individuals.
マウスの研究では、免疫によって誘発される胚中心反応のほとんどは 6 週間以内に解消されます;しかし、ヒトでは、Tfh 細胞と B 細胞の両方に対するナイーブ COVID-19 mRNA ワクチン反応は、多くの人で反応がピークに達する追加接種後 6 か月の時点でもまだ検出されています。
The kinetics of short-lived plasmablasts in the blood and of bone marrow-resident, long-lived plasma cells were also carefully measured.
血液中の短命の形質芽細胞と骨髄に存在する長命の形質細胞の動態も注意深く測定されました。
The rapid dynamic decay of the plasmablast response corresponds to measured declines in serum antibody.
形質芽細胞反応の急速な動的減衰は、測定された血清抗体の減少に対応します。
However, paired with the knowledge of ongoing germinal center function and output (as measured by increased numbers of Bmem cells), the maturation of improved and higher affinity antibodies is still occurring, despite waning total antibody levels.
しかし、進行中の胚中心の機能と生産量(Bmem細胞の数の増加によって測定される)に関する知識と組み合わせると、総抗体レベルが低下しているにもかかわらず、改善されたより高い親和性の抗体の成熟が依然として発生しています。
The rapid evolution of SARS-CoV-2 led to escape from the neutralizing antibody responses raised against the ancestral vaccine antigens.
SARS-CoV-2 の急速な進化により、祖先ワクチン抗原に対して引き起こされる中和抗体反応から逃れることができました。
However, CD4+ and CD8+ T cell responses are well characterized for these vaccines, and there is strong evidence that T cells play an important role in limiting serious infection-associated pathology.
ただし、これらのワクチンの CD4+ および CD8+ T 細胞応答はよく特徴付けられており、T 細胞が重篤な感染関連の病状を制限する上で重要な役割を果たしているという強力な証拠があります。
These data were derived from both animal studies and from humans who were on treatments that suppressed the B cell response.
これらのデータは、動物実験と、B 細胞反応を抑制する治療を受けたヒトの両方から得られたものです。
A greater understanding of the quantitative contribution of T cell responses to human vaccine protection will aid future vaccine design programs.
ヒトのワクチン防御に対する T 細胞応答の定量的寄与をより深く理解することは、将来のワクチン設計プログラムに役立ちます。
Inducing potent immune memory by vaccination requires a balance between activating robust lymphocyte responses and causing inflammatory sequelae.
ワクチン接種によって強力な免疫記憶を誘導するには、強力なリンパ球反応の活性化と炎症性続発症の原因との間のバランスが必要です。
This balancing act can be achieved by adjuvants, “the immunologist’s dirty little secret” described by Charles Janeway before mechanisms of action for vaccines were understood.
このバランスをとる作用は、ワクチンの作用機序が理解される前にチャールズ・ジェインウェイによって説明された「免疫学者のちょっとした秘密」であるアジュバントによって達成できます。
In modern vaccines, adjuvants that activate a single innate immune pathway (e.g., specific Toll-like receptors) are being tested for their capacity to prime a proper adaptive immune response.
最新のワクチンでは、単一の自然免疫経路(例:特定のToll様受容体)を活性化するアジュバントが、適切な適応免疫応答を刺激する能力についてテストされています。
Further, modern vaccine design also focuses on driving specific forms of adaptive immune memory.
さらに、最新のワクチン設計は、特定の形態の適応免疫記憶を促進することにも焦点を当てています。
For example, the generation of both CD4+ and CD8+ TRM cells is a major priority of next-generation vaccine initiatives, with mucosal vaccine platforms, such as inactivated viruses and inhaled vaccines in the airways, being tested.
たとえば、CD4+ および CD8+ TRM 細胞の生成は次世代ワクチン構想の主要な優先事項であり、不活化ウイルスや気道の吸入ワクチンなどの粘膜ワクチン プラットフォームがテストされています。
The goal is to localize lymphocyte memory into the upper or lower airways to more faithfully mirror the potent TRM responses generated by a pathogenic infection of the respiratory system.
目標は、リンパ球の記憶を上気道または下気道に局在させ、呼吸器系の病原性感染によって生じる強力な TRM 応答をより忠実に反映することです。
However, as with pathogenic infection, vaccine-induced inflammation in the airways, lung, intestinal, or genital mucosa via a vaccination needs to be carefully regulated to limit any immunopathology.
ただし、病原体感染と同様、ワクチン接種による気道、肺、腸管、または生殖器粘膜におけるワクチン誘発性の炎症は、免疫病理を制限するために慎重に制御する必要があります。
Beyond infectious diseases, there are rapidly growing efforts in vaccination against tumor antigens, including unmutated tumor-associated antigens and personalized or public neoantigens.
感染症を超えて、非変異腫瘍関連抗原や個別化または公開ネオアンチゲンなどの腫瘍抗原に対するワクチン接種の取り組みが急速に増えています。
For most tumor vaccination, the immunization occurs after the tumor has formed and seeks to elicit cytotoxic CD4+ and CD8+ T cell responses against the tumor.
ほとんどの腫瘍ワクチン接種では、腫瘍が形成された後に免疫化が行われ、腫瘍に対する細胞傷害性 CD4+ および CD8+ T 細胞応答を誘発しようとします。
Immunizing against tumor antigens presents complications due to the close or identical relationship of the target antigen to self-antigens and the unforgiving environment of the tumor and local lymphatics.
腫瘍抗原に対する免疫化は、標的抗原と自己抗原の密接または同一の関係、および腫瘍および局所リンパ管の過酷な環境により、合併症を引き起こします。
As such, the tumor microenvironment can limit T cell activity by suppressing function and limiting migration.
したがって、腫瘍微小環境は、機能を抑制し、遊走を制限することにより、T 細胞の活性を制限する可能性があります。
However, recent tumor vaccination efforts have shown significant promise in solid tumors, including in pancreatic cancer and melanoma.
しかし、最近の腫瘍ワクチン接種の取り組みは、膵臓がんや黒色腫などの固形腫瘍において大きな期待を示しています。
In addition, certain tumors are promoted by viral infection, including the human papillomavirus (HPV), which plays a causative role in the development of many cervical and head and neck cancers.
さらに、特定の腫瘍は、多くの子宮頸がんや頭頸部がんの発症の原因となるヒトパピローマウイルス(HPV)などのウイルス感染によって促進されます。
The introduction of HPV vaccines has profoundly lowered the risk of cancer in recipient populations, demonstrating the potential efficacy for preventive anti-cancer vaccines for tumors where there are established, shared antigens.
HPV ワクチンの導入により、レシピエント集団におけるがんのリスクが大幅に低下し、確立された共有抗原が存在する腫瘍に対する予防的抗がんワクチンの潜在的な有効性が実証されました。
In combination with other immunotherapeutic approaches, tumor vaccines are an important area for future mechanistic investigation.
他の免疫療法アプローチと組み合わせると、腫瘍ワクチンは将来のメカニズム研究にとって重要な分野となります。
T cells as living drugs
生きた薬としてのT細胞
T cell-based cell therapies are built upon decades of basic research.
T 細胞ベースの細胞療法は、数十年にわたる基礎研究に基づいて構築されています。
The convergence of immunobiology and synthetic biology (called synthetic immunology) has broadened the applicability of cell therapies, especially by pioneering the use of genetically reprogrammed T cells to treat tumors, infectious disease, and autoimmunity.
免疫生物学と合成生物学 の融合 (合成免疫学と呼ばれる)により、特に腫瘍、感染症、自己免疫の治療における遺伝子的に再プログラムされた T 細胞の使用の先駆者となり、細胞療法の適用可能性が広がりました。
Cellular therapies for cancer include use of T cells that are expanded after isolation from the tumor site (TILs), chimeric antigen receptor T cells (CAR-T), and TCR-engineered T cells (TCR-T) (Figure 6A).
がんの細胞療法には、腫瘍部位から単離した後に増殖する T 細胞 (TIL)、キメラ抗原受容体 T 細胞 (CAR-T)、および TCR 改変 T 細胞 (TCR-T) の使用が含まれます (図 6A)。
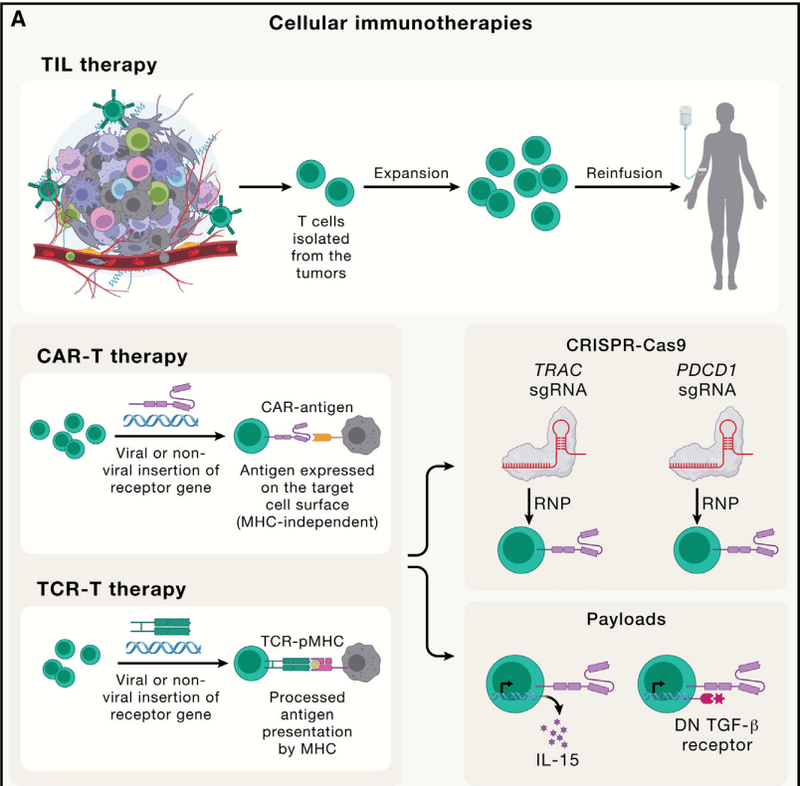
TIL therapy has shown promising efficacy and was approved by the US Food and Drug Administration (FDA) in 2024 to treat melanoma (Amtagvi).
TIL 療法は有望な有効性を示しており、2024 年に米国食品医薬品局 (FDA) によって黒色腫 (Amtagvi) の治療薬として承認されました。
However, isolation and preparation of abundant tumor-specific T cells are still difficult in other cancer types.
しかし、他の種類のがんでは、豊富な腫瘍特異的 T 細胞の単離と調製が依然として困難です。
CARs are fusion proteins that combine the antigen-binding domains of antibodies with T cell signaling domains and recognize cancer cell surface antigens independently of MHC-mediated peptide presentation.
CAR は、抗体の抗原結合ドメインと T 細胞シグナル伝達ドメインを組み合わせた融合タンパク質で、MHC 媒介ペプチド提示とは独立して癌細胞表面抗原を認識します。
CAR-T therapy has achieved strong clinical outcomes in treating B cell malignancies, leading to FDA approval of CD19-targeting CARs to treat B cell leukemias and lymphomas.
CAR-T 療法は、B 細胞悪性腫瘍の治療において強力な臨床成果を達成しています。 これにより、B細胞白血病およびリンパ腫を治療するためのCD19標的CARのFDA承認につながりました。
By contrast, CAR-T cells have shown limited efficacy toward solid tumors, whose immunosuppressive tumor microenvironment often causes CAR-T cell exclusion and exhaustion.
対照的に、CAR-T 細胞は固形腫瘍に対して限定的な有効性を示しています。 その免疫抑制性腫瘍微小環境は、CAR-T 細胞の排除と枯渇を引き起こすことがよくあります。
Also, it remains challenging to identify target antigens with high-level and homogenous expression on solid tumors but not normal tissues.
また、固形腫瘍では高レベルかつ均一に発現するが正常組織では発現しない標的抗原を同定することは依然として困難である。
Logic-gated CAR-T cells, which express two different CARs on their surface and must sense two separate antigens for activation, may overcome this obstacle.
論理ゲート型 CAR-T 細胞は、この障害を克服できる可能性があります。 論理ゲート型 CAR-T 細胞は、表面に 2 つの異なる CAR を発現し、活性化のために 2 つの異なる抗原を感知する必要があります。
Finally, TCR-T therapy uses an ectopically expressed TCR recognizing a tumor antigen to expand tumor-specific T cells for enhanced killing activity.
最後に、TCR-T 療法では、腫瘍抗原を認識する異所的に発現した TCR を使用して、腫瘍特異的 T 細胞を増殖させ、殺傷活性を高めます。
TCR-T cells are subject to MHC restriction and thus will only function in individuals with that MHC.
TCR-T 細胞は MHC 制限を受けるため、その MHC を持つ個体でのみ機能します。
New strategies to modify intracellular metabolic, proliferation, and survival pathways can further boost the efficacies of these cell therapies.
細胞内の代謝、増殖、生存経路を変更する新しい戦略により、これらの細胞療法の有効性をさらに高めることができます。
Moreover, CRISPR technology has advanced genetic engineering to improve T cell potency by offering applications such as unbiased functional screens and site-specific genetic modifications.
さらに、CRISPR テクノロジーは、偏りのない機能スクリーニングや部位特異的遺伝子修飾などのアプリケーションを提供することにより、T 細胞の効力を向上させる遺伝子工学を進歩させました。
Recent in vivo or multimodal CRISPR screens in T cells identified new targets that, when deleted, reprogram cells to boost CAR-T cell function.
最近の T 細胞における in vivo またはマルチモーダル CRISPR スクリーニングでは、削除されると細胞を再プログラムして CAR-T 細胞の機能を高める新しい標的が同定されました。
In vitro CRISPR screens uncovered that IFNγR signaling is required for CAR binding duration and avidity in solid but not liquid tumors.
in vitro CRISPR スクリーニングにより、液体腫瘍ではなく固形腫瘍における CAR 結合持続時間と結合力に IFNγR シグナル伝達が必要であることが明らかになりました。
Also, inserting CAR expression into specific genetic loci (e.g., TRAC or PDCD1) through CRISPR technology further improves CAR-T efficiency.
また、CRISPR テクノロジーを通じて CAR 発現を特定の遺伝子座 (TRAC または PDCD1 など) に挿入すると、CAR-T 効率がさらに向上します。
Finally, engineered T cells can act as vehicles to deliver therapeutic “payloads” to improve effector function.
最後に、改変された T 細胞は、治療用の「ペイロード」を送達する媒体として機能し、エフェクター機能を向上させることができます。
These payloads include cytokines (e.g., IL-15 and IL-10 to enhance persistence and resistance to dysfunction, respectively), dominant-negative receptors as a “sink” for immunosuppressive cytokines (e.g., TGF-β), or antibodies (e.g., anti-PD-1 to overcome exhaustion).
これらのペイロードには、免疫抑制性サイトカイン(例:TGF-β)または抗体(例:疲労を克服するための抗PD-1)の「シンク」として、サイトカイン (たとえば、機能不全に対する持続性と抵抗性をそれぞれ強化する IL-15 と IL-10)、ドミナントネガティブ受容体が含まれます。
Beyond cancer, T cell-based cell therapies are emerging for infectious disease and other immune-mediated diseases, such as engineering of CAR-Treg cells to treat autoimmune diseases and transplantation.
がん以外にも、自己免疫疾患や移植を治療するための CAR-Treg 細胞の操作など、感染症やその他の免疫介在性疾患に対する T 細胞ベースの細胞療法が登場しています。
Opportunities and challenges remain for these living drugs in the future for these non-malignant diseases.
これらの非悪性疾患に対するこれらの生薬には、将来的にも機会と課題が残されています。
Antibodies as therapies
治療法としての抗体
Due to their high specificity for target antigens, mAbs represent a major category of therapies for cancer, inflammation, and other diseases (Figure 6B).
標的抗原に対する特異性が高いため、mAb は癌、炎症、その他の疾患の主要な治療法の代表となります (図 6B)。
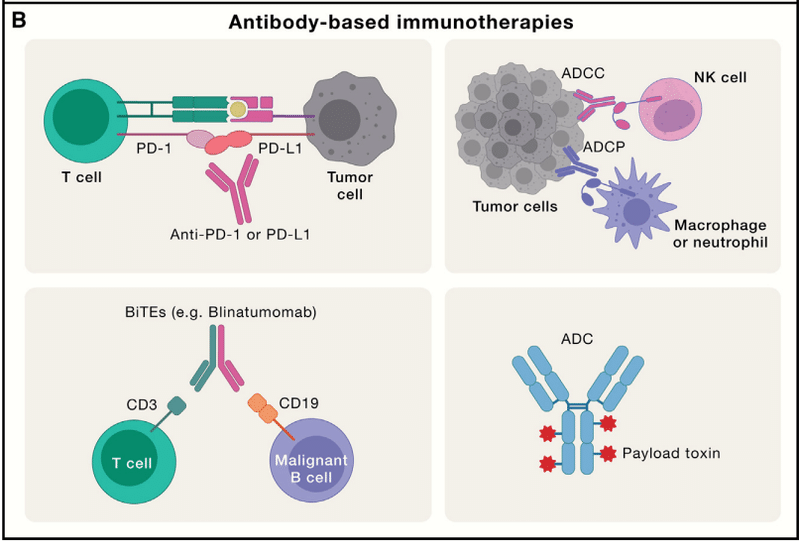
Following the approval of the first anti-CD3 mAb therapy for transplant rejection (muromonab, more commonly called OKT3) in 1986, antibody-based therapies have rapidly emerged with remarkable clinical and commercial success.
1986 年に移植拒絶反応に対する最初の抗 CD3 mAb 療法 (ムロモナブ、より一般的には OKT3 と呼ばれる) が承認されてから、抗体ベースの療法が急速に出現し、臨床的および商業的に目覚ましい成功を収めました。
Further, mAb engineering has evolved from the first generation (mouse antibodies with names ending in -omab) to the second (human-mouse chimera, -ximab), the third (humanized, -zumab), and finally the fourth (fully human, -umab) generation, each associated with diverse mechanisms of action.
さらに、mAb エンジニアリングは、第 1 世代 (名前が -omab で終わるマウス抗体) から、第 2 世代 (ヒト-マウスのキメラ、-ximab)、第 3 世代 (ヒト化、-zumab) へと進化してきました。 そして最後に第 4 世代 (完全なヒト、-umab) があり、それぞれが多様な作用機序に関連付けられています。
By targeting co-inhibitory molecules such as CTLA4 and PD-1 (or its ligand PD-L1) via therapeutic antibodies, ICB therapy directly unleashes endogenous T cell responses to target cancer.
ICB 療法は、治療用抗体を介して CTLA4 や PD-1 (またはそのリガンド PD-L1) などの共阻害分子を標的にすることで、標的がんに対する内因性 T 細胞応答を直接解き放ちます。
Though many patients experience durable tumor regression, most patients are unresponsive or develop resistance to current forms of ICB.
多くの患者は永続的な腫瘍退縮を経験しますが、ほとんどの患者は反応しないか、現在の形態の ICB に対して耐性を発現します。
Predicting the response to ICB remains challenging, although the composition or gene expression profiles of immune cells in tumors and the periphery serve as the most pertinent markers.
ICB に対する反応を予測することは依然として困難ですが、腫瘍および末梢における免疫細胞の組成または遺伝子発現プロファイルが最も適切なマーカーとして機能します。
Beyond the cancer context, ICB immunotherapies are being tested to treat malaria and HIV infection.
がんの枠を超えて、ICB 免疫療法はマラリアや HIV 感染症の治療のために試験されています。
Other antibodies that block ligand-receptor interactions are highly efficacious in the clinic, as exemplified by blocking cytokines (e.g., TNF-α by adalimumab, IL-4Rα by dupilumab, and IL-17 by secukinumab) that have transformed the treatment of autoimmune and allergic diseases.
リガンドと受容体の相互作用をブロックする他の抗体は臨床で非常に有効です。 代表例として、自己免疫疾患やアレルギー疾患の治療法を変えたサイトカイン(例:アダリムマブによるTNF-α、デュピルマブによるIL-4Rα、セクキヌマブによるIL-17)のブロックがあります。
Besides targeting ligand-receptor interactions, these therapeutic antibodies direct cell-mediated cytotoxicity toward tumors.
これらの治療用抗体は、リガンドと受容体の相互作用を標的とすることに加えて、細胞媒介細胞毒性を腫瘍に向けます。
For instance, anti-CD20 mAbs may induce caspase-independent programmed cell death of malignant B cells, whereas antibodies targeting death receptors cause caspase-dependent apoptosis.
たとえば、抗 CD20 mAb は悪性 B 細胞のカスパーゼ非依存性プログラム細胞死を誘導する可能性があります。 一方、デスレセプターを標的とする抗体はカスパーゼ依存性のアポトーシスを引き起こします。
Moreover, antibodies elicit cell-mediated immune reactions, whereby other immune cells (namely, natural killer [NK] cells and phagocytes) target and kill antigen-expressing target cells bound by the therapeutic antibodies.
さらに、抗体は細胞媒介性免疫反応を誘発し、それによって他の免疫細胞(すなわち、ナチュラルキラー [NK] 細胞および食細胞)が治療用抗体が結合した抗原発現標的細胞を標的として殺します。
Compared with mAbs using full-length IgG antibodies, the bispecific T cell engager (BiTE) is composed of an antibody fragment for activating the TCR-CD3 chain and a tumor-specific antigen to bridge the interaction between T cells and target cancer cells, thereby inducing an artificial immune synapse that promotes T cell-mediated killing of target cells independent of TCR specificity.
完全長 IgG 抗体を使用する mAb と比較して、二重特異性 T セルエンゲージャー (BiTE) は、TCR-CD3 鎖を活性化する抗体フラグメントと、T 細胞と標的がん細胞の間の相互作用を橋渡しする腫瘍特異的抗原で構成されています。 これにより、TCR の特異性とは無関係に、T 細胞を介した標的細胞の死滅を促進する人工免疫シナプスが誘導されます。
Finally, there is a growing interest in antibody-drug conjugates (ADCs) composed of a tumor antigen-targeting antibody, a linker, and a payload toxin (tubulin-binding or DNA-targeting agents).
最後に、腫瘍抗原を標的とする抗体、リンカー、およびペイロード毒素 (チューブリン結合剤または DNA 標的剤) で構成される抗体薬物複合体 (ADC) への関心が高まっています。
After the ADC is internalized by target tumor cells, the linker is cleaved to release the toxin.
ADC が標的腫瘍細胞に取り込まれた後、リンカーが切断されて毒素が放出されます。
Improving ADC-related efficacy while reducing side effects requires further optimization.
副作用を軽減しながら ADC 関連の有効性を改善するには、さらなる最適化が必要です。
Aside from the direct use of T cells and antibodies as therapies, cytokine-based therapies, such as small-molecule inhibitors of JAK, have achieved clinical success in autoimmunity and inflammation.
治療法としての T 細胞と抗体の直接使用とは別に、JAK の小分子阻害剤などのサイトカインベースの治療法は、自己免疫と炎症において臨床的な成功を収めています。
Whereas direct use of cytokines has shown limited effects, possibly due to their pleiotropic effects and rapid diffusion, innovations in bioengineering have recently yielded promising new technologies in modulating adaptive immunity.
サイトカインの直接使用は、おそらくその多面発現効果と急速な拡散のため、効果が限られていることが示されていますが、最近、生物工学の革新により、適応免疫を調節する有望な新技術が生み出されています。
For example, although IL-2 drives both expansion and exhaustion of CD8+ T cells, a partial agonist of IL-2 retains the ability to induce expansion and better maintain the TCF-1+ stem-like phenotype to escape exhaustion.
たとえば、IL-2 は CD8+ T 細胞の増殖と枯渇の両方を促進しますが、IL-2 の部分アゴニストは増殖を誘導し、枯渇から逃れるために TCF-1+ ステム様表現型をより良く維持する能力を保持しています。
Overall, limited efficacy remains a major hurdle of these immune-based therapies, and combination therapies are key to overcoming therapeutic resistance.
全体として、限定された有効性がこれらの免疫ベースの治療の大きな障害となっており、併用療法が治療抵抗性を克服する鍵となります。
Concluding remarks
Here, we have highlighted the core and emerging principles of adaptive immunity and its applications to immunotherapy.
ここでは、適応免疫の中核となる原理と新たな原理、およびその免疫療法への応用に焦点を当てました。
Since the discoveries of T cells and B cells in the 1960s, exploring adaptive immunity has yielded pivotal contributions to immunology.
1960 年代に T 細胞と B 細胞が発見されて以来、適応免疫の探求は免疫学に極めて重要な貢献をもたらしてきました。
Adaptive immune cells are also an excellent system to study fundamentals of cell, molecular, and structural biology and mechanisms of metabolic, signaling, epigenetic, and transcriptional regulation, leading to major discoveries beyond immunology.
適応免疫細胞は、細胞生物学、分子生物学、構造生物学の基礎と、代謝、シグナル伝達、エピジェネティック、転写制御のメカニズムを研究するための優れたシステムでもあり、免疫学を超えた重要な発見につながります。
For instance, investigating immunometabolism at the signaling, cellular, and systemic (e.g., dietary interventions and microbiota-derived nutrients) levels has enriched our knowledge on immunity and metabolism and provided new opportunities for targeting cancer, metabolic disorders, and other immune-mediated diseases.
たとえば、免疫代謝をシグナル伝達、細胞、全身(食事介入や微生物叢由来の栄養素など)レベルで研究することで、免疫と代謝に関する知識が豊富になりました。 そして、がん、代謝障害、その他の免疫介在性疾患を標的とする新たな機会を提供しました。
Similarly, investigation of epigenetic and chromatin regulation in lymphocytes has generated new concepts in context-specific gene regulation.
同様に、リンパ球におけるエピジェネティックな制御およびクロマチン制御の研究により、状況特異的な遺伝子制御における新しい概念が生み出されました。
Continued exploration of adaptive immunity and its intersection with fundamental biology will advance our understanding of the principles that are unique to the immune system and those that are broadly applicable to other biological systems.
適応免疫とその基礎生物学との関わりの継続的な探求により、免疫系に特有の原理と他の生物学的システムに広く適用できる原理の理解が進むでしょう。
Tissue immunity is an emerging regulator of physiology and disease.
組織免疫は、生理機能と病気の新たな制御因子です。
Unlike other cells, adaptive immune cells face the unique challenges of entering and acclimating to varying environmental cues in diverse tissues.
他の細胞とは異なり、適応免疫細胞は、さまざまな組織のさまざまな環境の合図に入り、それに順応するという独特の課題に直面しています。
Tissue context-specific adaptation is therefore fundamental to understanding tissue physiology and disease pathogenesis; however, much remains to be learned about the impact of the diverse tissue microenvironments on the adaptation of immune cells, and vice versa, as well as the complex immune-tissue cell crosstalk.
したがって、組織の状況に特異的な適応は、組織の生理学と疾患の病因を理解するための基礎となります;しかし、多様な組織微小環境が免疫細胞の適応に及ぼす影響、およびその逆の影響や、複雑な免疫組織細胞のクロストークについては、まだ解明されていないことが多くあります。
To that end, recent interrogations of adaptive immunity in non-lymphoid tissues have markedly advanced our knowledge of tissue immunity (e.g., tissue-resident cells and Treg-mediated tissue repair).
この目的を達成するために、非リンパ系組織における適応免疫に関する最近の調査により、組織免疫(例えば、組織常在細胞およびTreg媒介組織修復)に関する知識が著しく進歩した。
More importantly, the integration of adaptive immunity with physiology leads to an in-depth understanding of normal and diseased states of various tissues and the organism.
さらに重要なことは、適応免疫と生理学を統合することで、さまざまな組織や生体の正常な状態と病気の状態についての深い理解につながるということです。
For instance, the information on tumor-immune or neuroimmune interactions has contributed to therapeutic interventions of cancer and other diseases.
たとえば、腫瘍と免疫または神経免疫の相互作用に関する情報は、がんやその他の疾患の治療介入に貢献しています。
However, we are only beginning to understand how the immune system shapes tissue homeostasis or dysregulation in aging-related, neurodegenerative, or metabolic disorders.
しかし、免疫系が老化関連疾患、神経変性疾患、または代謝性疾患における組織の恒常性や調節不全をどのように形成するのか、私たちは理解し始めたばかりです。
How adaptive immunity, including host-intrinsic (e.g., tissue-specific immunity) and extrinsic (e.g., infection, microbiota, or nutrition) factors, intersects with normal physiology and disease states will likely bring fundamental new insights into immunobiology, with remarkable translational and therapeutic potentials.
宿主内因性(例:組織特異的免疫)および外因性(例:感染、微生物叢、または栄養)因子を含む適応免疫が、正常な生理機能および疾患状態とどのように交差するかは、おそらく、免疫生物学に根本的な新しい洞察をもたらし、顕著な翻訳および治療の可能性をもたらすでしょう。
Immunology is at the forefront of technology development and innovation, with ever-increasing resolution and ever-expanding dimensions to probe the complex adaptive immune system.
免疫学は技術開発と革新の最前線にあり、複雑な適応免疫システムを調査するための解像度が高まり、次元が拡大し続けています。
Historically, the application of flow cytometry permitted single-cell analysis of immune responses, which was instrumental for discovery and analysis of lymphocyte subsets, whereas mouse conditional genetic models allowed for cell type-specific or spatiotemporally controlled modulation of gene function in vivo.
歴史的に、フローサイトメトリーの応用により免疫応答の単一細胞分析が可能になり、これはリンパ球サブセットの発見と分析に役立ちました。 一方、マウスの条件付き遺伝モデルでは、in vivo での遺伝子機能の細胞型特異的または時空間的に制御された調節が可能になりました。
Recent innovative technologies have provided unprecedented opportunities for high-throughput analysis of adaptive immune responses.
最近の革新的なテクノロジーにより、適応免疫応答のハイスループット分析の前例のない機会が提供されています。
For instance, the use of scRNA-seq permits the measurement of thousands of transcripts rather than limited markers offered by flow cytometry and can be integrated with protein expressional data and spatially resolved information (e.g., via spatial transcriptomics).
例えば、scRNA-seq を使用すると、フローサイトメトリーによって提供される限られたマーカーではなく、数千の転写産物の測定が可能になり、タンパク質発現データや空間的に分解された情報(例えば、空間トランスクリプトミクスによる)と統合することができます。
Further, CRISPR-based pooled screens in vivo enable the unbiased discovery of the most functionally relevant targets under physiologically relevant conditions.
さらに、CRISPR ベースの in vivo プールスクリーニングにより、生理学的に関連する条件下で最も機能的に関連する標的を公平に発見することが可能になります。
Similarly, we have moved from analysis of individual antigen receptors to unbiased profiling of the entire TCR or BCR repertoire at single-cell resolution.
同様に、我々は個々の抗原受容体の分析から、単一細胞解像度での TCR または BCR レパートリー全体の不偏プロファイリングに移行しました。
The integrative use of these multi-omics tools requires expertise in computational biology and systems biology to uncover biological insights and disease targets.
これらのマルチオミクスツールを統合的に使用するには、生物学的洞察と疾患標的を明らかにするために、計算生物学とシステム生物学の専門知識が必要です。
Therefore, systems immunology serves as a primary example of successful integration of experimental biology and data science for biological and clinical discovery.
したがって、システム免疫学は、生物学的および臨床的発見のための実験生物学とデータサイエンスの統合に成功した主な例として機能します。
The synthesis of experimental and computational biology and integration of immunobiology, cutting-edge technologies, and data science will drive the next-generation research in adaptive immunity and produce new innovations.
実験生物学と計算生物学の統合、免疫生物学、最先端技術、データサイエンスの統合は、適応免疫における次世代の研究を推進し、新たなイノベーションを生み出すでしょう。
Adaptive immunity may also hold the key to understanding and curing a broad spectrum of human diseases.
適応免疫は、広範囲にわたる人間の病気を理解し、治療するための鍵を握る可能性もあります。
This notion is best exemplified by the successes in vaccine development for infectious disease, immunotherapies for cancer, and cytokine-targeting therapies for inflammatory diseases.
この概念は、感染症のワクチン開発、癌の免疫療法、炎症性疾患のサイトカイン標的療法の成功によって最もよく例証されています。
Indeed, basic research and fundamental discoveries using animal models (e.g., immune checkpoint molecules, inflammatory cytokines, and Th17 cells) have laid the foundation for innovative immunotherapies and clinical translation.
実際、動物モデルを使用した基礎研究と基礎的発見(免疫チェックポイント分子、炎症性サイトカイン、Th17 細胞など)は、革新的な免疫療法と臨床応用の基礎を築きました。
Aside from this traditional bench-to-bedside translation, bedside-to-bench is being increasingly applied.
この従来のベンチからベッドサイドへの変換とは別に、ベッドサイドからベンチへの変換がますます適用されています。
In particular, with the advances in high-throughput technologies (e.g., scRNA-seq, spatial transcriptomics, and multiplexed imaging), the use of human patient-derived materials has moved beyond observational phenotyping to in-depth mechanistic investigation of disease state and precision medicine, accelerated by complementary model systems, such as organoids and advanced mouse genetic models (e.g., humanized mice).
特に、ハイスループット技術 (scRNA-seq、空間トランスクリプトミクス、多重化イメージングなど) の進歩により、 ヒト患者由来の材料の使用は、観察的な表現型解析を超えて、オルガノイドや高度なマウス遺伝子モデル(ヒト化マウスなど)などの相補的モデルシステムによって加速され、病状の詳細な機構研究や精密医療へと移行しています。
Moreover, immune engineering, as evidenced by the remarkable success of CAR-T living drugs and antibody and cytokine engineering, will continue to drive innovation and new opportunities for translation.
さらに、CAR-T生薬や抗体、サイトカイン工学の目覚ましい成功によって証明されているように、免疫工学は今後も革新と新たな翻訳の機会を推進していくだろう。
In summary, the intersection of adaptive immunity with biology, physiology, technology, and therapy has produced major breakthroughs and transformed biological and clinical sciences, and we are undoubtedly on the brink of many more breakthroughs in the next half century.
要約すると、適応免疫と生物学、生理学、テクノロジー、治療法との交差点は大きな進歩をもたらし、生物学と臨床科学に変革をもたらしました。そして、私たちは間違いなく、今後半世紀でさらに多くの画期的な進歩の間際にいます。
以下省略。
この記事が気に入ったらサポートをしてみませんか?