COVID-19 における宿主プロテアーゼの役割と、その機能を薬理学的に制御することの重要性を理解

Aprotinin (I): Understanding the Role of Host Proteases in COVID-19 and the Importance of Pharmacologically Regulating Their Function
アプロチニン (I): COVID-19 における宿主プロテアーゼの役割と、その機能を薬理学的に制御することの重要性を理解する
Abstract
Proteases are produced and released in the mucosal cells of the respiratory tract and have important physiological functions, for example, maintaining airway humidification to allow proper gas exchange.
プロテアーゼは呼吸器の粘膜細胞で生成され、放出され、例えば気道の加湿を維持して適切なガス交換を可能にするなど、重要な生理学的機能を持っています。
The infectious mechanism of severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), takes advantage of host proteases in two ways: to change the spatial conformation of the spike (S) protein via endoproteolysis (e.g., transmembrane serine protease type 2 (TMPRSS2)) and as a target to anchor to epithelial cells (e.g., angiotensin-converting enzyme 2 (ACE2)).
コロナウイルス疾患 2019 (COVID-19) を引き起こす重症急性呼吸器症候群コロナウイルス 2 型 (SARS-CoV-2) の感染メカニズムは、2 つの方法で宿主プロテアーゼを利用します。エンドプロテアーゼ分解を介してスパイク (S) タンパク質の空間的構造を変更すること (例: 膜貫通型セリンプロテアーゼ 2 型 (TMPRSS2)) と、上皮細胞に固定するターゲットとして利用すること (例: アンジオテンシン変換酵素 2 (ACE2)) です。
This infectious process leads to an imbalance in the mucosa between the release and action of proteases versus regulation by anti-proteases, which contributes to the exacerbation of the inflammatory and prothrombotic response in COVID-19.
この感染プロセスにより、粘膜におけるプロテアーゼの放出と作用と抗プロテアーゼによる調節との間の不均衡が生じ、COVID-19 における炎症および血栓形成反応の悪化に寄与します。
In this article, we describe the most important proteases that are affected in COVID-19, and how their overactivation affects the three main physiological systems in which they participate: the complement system and the kinin–kallikrein system (KKS), which both form part of the contact system of innate immunity, and the renin–angiotensin–aldosterone system (RAAS).
この記事では、COVID-19 で影響を受ける最も重要なプロテアーゼについて説明し、それらの過剰活性化が、それらが関与する 3 つの主要な生理学的システム、すなわち自然免疫の接触システムの一部を形成する補体系とキニン-カリクレイン系 (KKS)、およびレニン-アンジオテンシン-アルドステロン系 (RAAS) にどのように影響するかについて説明します。
We aim to elucidate the pathophysiological bases of COVID-19 in the context of the imbalance between the action of proteases and anti-proteases to understand the mechanism of aprotinin action (a panprotease inhibitor).
私たちは、プロテアーゼと抗プロテアーゼの作用の不均衡という文脈で COVID-19 の病態生理学的根拠を解明し、アプロチニン (汎プロテアーゼ阻害剤) の作用メカニズムを理解することを目指しています。
In a second-part review, titled “Aprotinin (II): Inhalational Administration for the Treatment of COVID-19 and Other Viral Conditions”, we explain in depth the pharmacodynamics, pharmacokinetics, toxicity, and use of aprotinin as an antiviral drug.
「アプロチニン(II):COVID-19およびその他のウイルス性疾患の治療のための吸入投与」と題された第2部のレビューでは、抗ウイルス薬としてのアプロチニンの薬力学、薬物動態、毒性、および使用について詳しく説明します。
1. Introduction
Coronaviruses infect epithelial cells by recognising and binding to certain plasma membrane proteins.
コロナウイルスは、特定の細胞膜タンパク質を認識して結合することで上皮細胞に感染します。
One of the most studied is the angiotensin-converting enzyme type 2 (ACE2).
最も研究されているものの1つは、アンジオテンシン変換酵素2型(ACE2)です。
The anchoring mechanism of severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), to this enzyme has consequences that are fundamental for the cell and is closely related to the disease it causes.
コロナウイルス疾患2019(COVID-19)を引き起こす重症急性呼吸器症候群コロナウイルス2型(SARS-CoV-2)のこの酵素への固定メカニズムは、細胞にとって基本的な結果をもたらし、それが引き起こす疾患と密接に関連しています。
To understand the pathophysiology COVID-19, it is necessary to explain the importance of ACE2, which goes beyond blood pressure control.
COVID-19の病態生理を理解するには、血圧制御を超えたACE2の重要性を説明する必要があります。
In this first review, titled “Aprotinin (I): Understanding the Role of Host Proteases in COVID-19 and the Importance of Pharmacologically Regulating Their Function”, we explain the pathophysiological importance of ACE in the SARS-CoV-2 infection process, and how a cascade of events activates proteolytic pathways that constitute the most important causes of the disease.
「アプロチニン(I):COVID-19における宿主プロテアーゼの役割とそれらの機能の薬理学的制御の重要性を理解する」と題されたこの最初のレビューでは、SARS-CoV-2感染プロセスにおけるACEの病態生理学的重要性と、一連のイベントがどのようにしてこの疾患の最も重要な原因を構成するタンパク質分解経路を活性化するかを説明します。
Understanding these mechanisms allows the development of new antiviral drugs, such as aprotinin, which is a broad-spectrum inhibitor of host proteases.
これらのメカニズムを理解することで、宿主プロテアーゼの広域スペクトル阻害剤であるアプロチニンなどの新しい抗ウイルス薬の開発が可能になります。
In the second-part review, titled “Aprotinin (II): Inhalational Administration for the Treatment of COVID-19 and Other Viral Conditions”, we describe the main pharmacodynamic, pharmacokinetic, and toxicological mechanisms of aprotinin for its use by inhalation in these conditions.
第 2 部のレビュー「アプロチニン (II): COVID-19 およびその他のウイルス性疾患の治療のための吸入投与」では、これらの疾患で吸入投与されるアプロチニンの主な薬力学的、薬物動態学的、および毒物学的メカニズムについて説明します。
2. The SARS-CoV-2 Infectious Process
SARS-CoV-2の感染プロセス
ACE2, the main target to which SARS-CoV-2 anchors in the viral infectious process, is expressed to a greater or lesser extent in the cells of the pulmonary, digestive, renal, and vascular endothelium.
ACE2は、SARS-CoV-2がウイルス感染プロセスで固定する主なターゲットであり、肺、消化管、腎臓、血管内皮の細胞で多かれ少なかれ発現しています。
Anchoring and fusion of the viral capsid to the host cell occurs through recognition of a virus envelope glycoprotein, known as the spike (S) protein, with this enzyme.
ウイルスカプシドの宿主細胞への固定と融合は、スパイク(S)タンパク質として知られるウイルスエンベロープ糖タンパク質をこの酵素で認識することによって起こります。
However, for the S protein to recognise it, it needs to undergo a post-translational activation process by endoproteolysis by proteases from the epithelial cells.
しかし、Sタンパク質がそれを認識するには、上皮細胞からのプロテアーゼによるエンドプロテアーゼ分解による翻訳後活性化プロセスを経る必要があります。
This two-step entry mechanism that involves activation by endogenous host proteases is common in viruses of the Paramyxoviridae, Orthomyxoviridae, Retroviridae, Herpesviridae, Flaviviridae, Filoviridae, Hepadnaviridae, Togaviridae, and Coronaviridae families.
内因性宿主プロテアーゼによる活性化を伴うこの2段階の侵入メカニズムは、パラミクソウイルス科、オルトミクソウイルス科、レトロウイルス科、ヘルペスウイルス科、フラビウイルス科、フィロウイルス科、ヘパドナウイルス科、トガウイルス科、およびコロナウイルス科のウイルスに共通しています。
In the specific case of SARS-CoV-2, the cleavage occurs in four redundant furin-like domains located in the S1/S2 protein subunits of the virus.
SARS-CoV-2 の特定のケースでは、ウイルスの S1/S2 タンパク質サブユニットにある 4 つの冗長なフーリン様ドメインで切断が起こります。
Proteolysis is indispensable to separate and activate the S1 and S2 subunits, each of which performs distinct functions (Figure 1A).
タンパク質分解は、それぞれ異なる機能を果たす S1 サブユニットと S2 サブユニットを分離して活性化するために不可欠です (図 1A)。
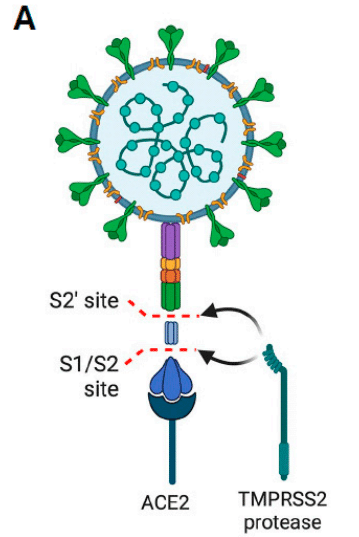
While the S1 subunit is responsible for binding to ACE2 with an affinity in the nanomolar range, the S2 subunit participates in the fusion between viral RNA and the cell membrane.
S1サブユニットはナノモル範囲の親和性でACE2に結合する役割を担う一方、S2サブユニットはウイルスRNAと細胞膜の融合に関与する。
Unlike other coronaviruses, this proteolytic cleavage and presentation of the S1 subunit for anchoring to ACE2 is much more efficient for SARS-CoV-2.
他のコロナウイルスとは異なり、このタンパク質分解による切断とS1サブユニットのACE2への固定のための提示は、SARS-CoV-2にとってはるかに効率的です。
Therefore, it is essential to know the host proteases used by SARS-CoV-2 for its infectious process to prevent disease.
したがって、SARS-CoV-2 が感染プロセスで使用する宿主プロテアーゼを知っておくことは、病気を予防するために不可欠です。
Among the known host proteases, trypsin, cysteine protease cathepsins, thermolysin, neutrophil elastase (NE), and activated clotting factors, such as plasminogen and factor Xa (FXa), have been described.
既知の宿主プロテアーゼとしては、トリプシン、システインプロテアーゼカテプシン、サーモリシン、好中球エラスターゼ (NE)、プラスミノーゲンや因子 Xa (FXa) などの活性化凝固因子が知られています。
In addition, much attention has been paid to transmembrane serine protease type 2 (TMPRSS2), one of the proteases for which SARS-CoV-2 shows the strongest preference to infect epithelial cells.
さらに、SARS-CoV-2が上皮細胞に感染する際に最も好んで用いるプロテアーゼの1つである膜貫通型セリンプロテアーゼ2型(TMPRSS2)にも注目が集まっている。
Once anchorage to ACE2 occurs, and depending on which protease is used in this process, the virus can enter the cell via at least two mechanisms.
ACE2への固定が起こると、このプロセスで使用されるプロテアーゼに応じて、ウイルスは少なくとも2つのメカニズムを介して細胞に侵入することができます。
If the S protein is cleaved via cathepsins, entry will occur through endosome formation.
Sタンパク質がカテプシンを介して切断される場合、侵入はエンドソーム形成を介して起こります。
In contrast, if proteolysis is via TMPRSS2, entry occurs through the formation of a fusion pore between its membrane and those of the epithelial cell.
対照的に、タンパク質分解がTMPRSS2を介して行われる場合、侵入は膜と上皮細胞の膜の間に融合孔を形成することによって起こります。
ACE2 is usually co-expressed with TMPRSS2, indicating the importance of this mechanism.
ACE2は通常TMPRSS2と共発現しており、このメカニズムの重要性を示しています。
However, the endocytic pathway usually occurs when TMPRSS2 expression levels are insufficient, and endosome formation occurs via a clathrin-dependent pathway, internalising the virus bound to ACE2.
しかし、エンドサイトーシス経路は通常、TMPRSS2の発現レベルが不十分な場合に発生し、クラスリン依存性経路を介してエンドソーム形成が発生し、ACE2に結合したウイルスが内部化されます。
The use of this endocytic pathway in respiratory cells that do not express TMPRSS2 explains why some SARS-CoV-2 variants, such as Omicron, have enhanced transmissibility.
TMPRSS2を発現しない呼吸器細胞でこのエンドサイトーシス経路が使用されることで、オミクロンなどの一部のSARS-CoV-2変異体の伝染性が高まった理由が説明されます。
Key points:
SARS-CoV-2 uses both soluble and membrane proteases of the host cell in its viral infection mechanism.
SARS-CoV-2 は、ウイルス感染メカニズムにおいて、宿主細胞の可溶性プロテアーゼと膜プロテアーゼの両方を使用します。
Depending on which protease you use will influence the way it enters the cell (e.g., endocytosis or through the formation of a fusion pore).
使用するプロテアーゼに応じて、細胞への侵入方法 (エンドサイトーシスまたは融合孔の形成など) が変わります。
2.1. The Physiological Importance of ACE beyond Blood Pressure Control
血圧コントロールを超えたACEの生理学的重要性
Although ACE was initially described for its importance in the control of blood pressure through the renin–angiotensin–aldosterone system (RAAS), it is now known to have relevance in other processes, such as renal embryonic development, reproduction, cell proliferation (e.g., haematopoiesis, myeloproliferation, and angiogenesis), inflammation, oxidative stress, and immunity.
ACE は当初、レニン-アンジオテンシン-アルドステロン系 (RAAS) を介した血圧制御における重要性について説明されていましたが、現在では腎臓の胚発生、生殖、細胞増殖 (造血、骨髄増殖、血管新生など)、炎症、酸化ストレス、免疫などの他のプロセスにも関連していることがわかっています。
In somatic tissues, ACE contains two catalytic domains, each with different affinities for protein substrates.
体細胞組織では、ACE には 2 つの触媒ドメインがあり、それぞれタンパク質基質に対する親和性が異なります。
While the carboxyl-terminal domain is specialised in cleaving angiotensin I to generate angiotensin II, the amino-terminal domain is also capable of cleaving other peptides, including enkephalin, the tetrapeptide N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), neurotensins, substance P, and bradykinin, among others (Figure 2) .
カルボキシル末端ドメインはアンジオテンシン I を切断してアンジオテンシン II を生成することに特化していますが、アミノ末端ドメインは、エンケファリン、テトラペプチド N-アセチルセリルアスパルチルリシルプロリン (AcSDKP)、ニューロテンシン、サブスタンス P、ブラジキニンなど、他のペプチドを切断することもできます (図 2) 。
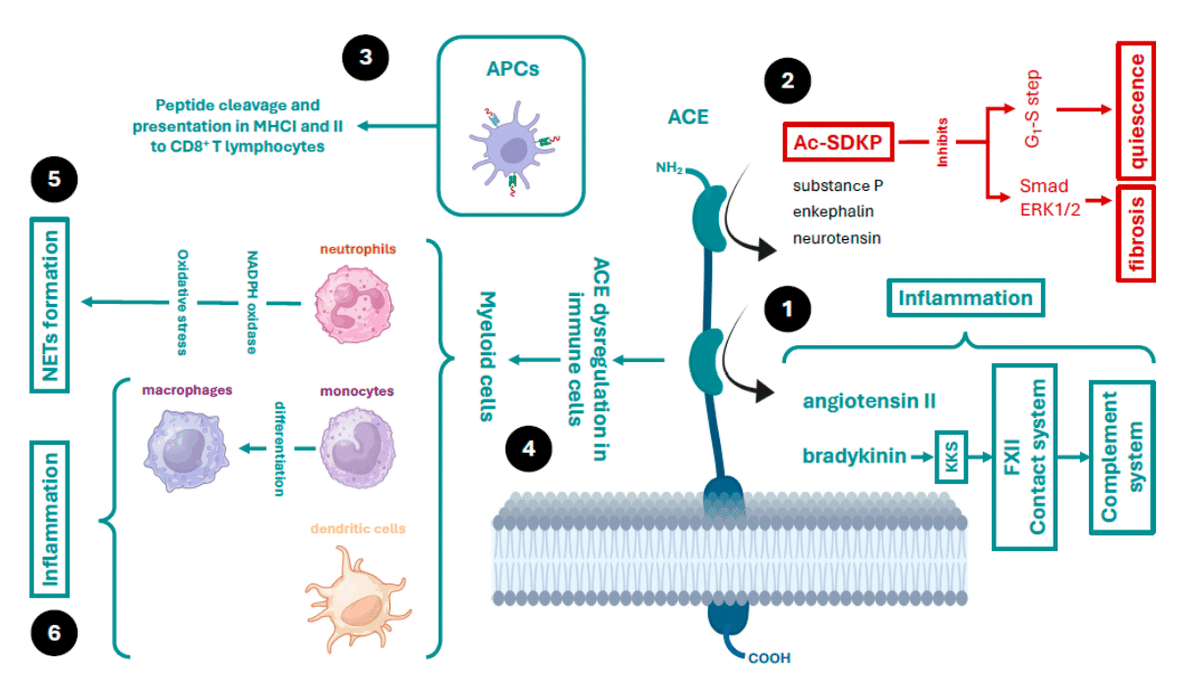
It can exert its protease function while anchored to the plasma membrane, extracellularly when released from cells into the blood plasma or subcellularly in cell organelles.
プロテアーゼ機能は、細胞膜に固定されている間、細胞外(細胞から血漿に放出されたとき)、または細胞小器官内の細胞内で発揮されます。
Thus, it is not only an endocrine, paracrine, and autocrine cellular communication system, it is also an intracellular communication system (between the cell organelles themselves).
したがって、これは内分泌、傍分泌、自己分泌の細胞コミュニケーションシステムであるだけでなく、細胞内コミュニケーションシステム(細胞小器官自体の間)でもあります。
These locations are important for the development of its various cellular functions and are of great relevance for understanding COVID-19 as well as the mechanisms by which SARS-CoV-2 evades the immune system.
これらの場所は、さまざまな細胞機能の発達に重要であり、COVID-19とSARS-CoV-2が免疫システムを回避するメカニズムを理解する上で非常に重要です。
ACE in Immunity
ACE is expressed in organelles, such as the endoplasmic reticulum, where it has been shown to be catalytically active, performing certain cellular functions.
ACE は小胞体などの細胞小器官で発現し、そこで触媒活性を示し、特定の細胞機能を果たすことが示されています。
Among these, it contributes to adaptive immunity by cleaving both endogenous and foreign peptides for presentation by antigen-presenting cells (macrophages and dendritic cells) via major histocompatibility complex type I (MHC-I) and II (MHC-II) to cluster of differentiation 8 (CD8)+ T cells (Figure 2).
これらのうち、ACE は、主要組織適合性複合体タイプ I (MHC-I) およびタイプ II (MHC-II) を介して抗原提示細胞 (マクロファージおよび樹状細胞) によって分化クラスター 8 (CD8)+ T 細胞に提示される内因性ペプチドと外来ペプチドの両方を切断することで、適応免疫に寄与します (図 2) 。
In addition, ACE can be overexpressed in myeloid cells, such as macrophages, neutrophils, and dendritic cells, thereby enhancing the innate immune response through the production of proinflammatory cytokines and nitric oxide.
さらに、ACE はマクロファージ、好中球、樹状細胞などの骨髄細胞で過剰発現し、炎症性サイトカインや一酸化窒素の産生を通じて自然免疫反応を増強する。
ACE plays a role in modulating the inflammatory response and recruitment of inflammatory cells, such as mast cells and neutrophils, by regulating the concentration of mediators and/or the expression of proteins, such as cytokines, adhesion molecules, and the plasma contact system, also known as the kinin–kallikrein system (KKS).
ACE は、サイトカイン、接着分子、キニン-カリクレイン系 (KKS) としても知られる血漿接触系などのメディエーターの濃度やタンパク質の発現を調節することにより、炎症反応やマスト細胞や好中球などの炎症細胞の動員を調整する役割を果たします。
ACE regulates this system by clearing the proinflammatory peptide bradykinin.
ACE は、炎症誘発性ペプチドであるブラジキニンを除去することにより、この系を制御します。
Increased bradykinin contributes to the activation of the FXII coagulation and complement pathways.
ブラジキニンの増加は、FXII 凝固経路および補体経路の活性化に寄与します。
ACE can be secreted into the extracellular environment (soluble ACE (sACE)) from these myeloid cells and act as a local or systemic regulator of the production of these peptides.
ACE はこれらの骨髄細胞から細胞外環境に分泌され (可溶性 ACE (sACE))、これらのペプチドの生成の局所的または全身的調節因子として作用します。
In neutrophils, ACE is essential for immune competence.
好中球では、ACE は免疫能に不可欠です。
These effects are independent of the angiotensin-converting action and depend on other factors—for example, activation of nicotinamide adenine dinucleotide phosphate oxidase (NOX) by ACE increases reactive oxygen species (ROS, including the superoxide) production.
これらの効果はアンジオテンシン変換作用とは無関係であり、他の要因に依存します。たとえば、ACE によるニコチンアミドアデニンジヌクレオチドリン酸オキシダーゼ (NOX) の活性化は、活性酸素種 (ROS、スーパーオキシドを含む) の生成を増加させます。
Neutrophils kill bacteria using phagocytic mechanisms and through the release of extracellular fibres, called neutrophil extracellular traps (NETs), which are in turn stimulated by ROS generation.
好中球は、貪食メカニズムと好中球細胞外トラップ(NET)と呼ばれる細胞外繊維の放出によって細菌を殺します。NETは、ROS生成によって刺激されます。
These fibres are composed of DNA and proteins that bind to, trap, and consequently kill bacteria.
これらの繊維は、細菌に結合してトラップし、その結果として細菌を殺す DNA とタンパク質で構成されています。
Moreover, under certain conditions, monocytes overexpress ACE, triggering further differentiation of these cells into macrophages, which release cytokines and adhesion and transmigration molecules.
さらに、特定の条件下では、単球が ACE を過剰発現し、これらの細胞がマクロファージにさらに分化して、サイトカインと接着分子および移行分子を放出します。
In addition, ACE is also involved in the production of nitric oxide, which is important for microbial defence by these cells (Figure 2).
さらに、ACE はこれらの細胞による微生物防御に重要な一酸化窒素の生成にも関与しています (図 2)。
Finally, ACE is involved in the control of immunity through the clearance of peptides with immunosuppressive activity, such as AcSDKP.
最後に、ACE は AcSDKP などの免疫抑制活性ペプチドの除去を通じて免疫制御に関与しています。
Among many actions, this peptide can inhibit the G1–S cell cycle transition and thus maintains haematopoietic progenitor cells in a quiescent phase.
このペプチドは多くの作用を持つが、G1-S細胞周期の移行を阻害し、造血前駆細胞を静止期に維持することができる。
This prevents proliferation, migration, and cytokine release by myeloid cells.
これにより、骨髄細胞の増殖、遊走、サイトカインの放出が防止されます。
In addition, it can prevent fibroblast collagen synthesis and deposition by inhibiting DNA synthesis and endothelin-1, and by blocking small mothers against decapentaplegic (Smad) signal transduction and the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, thus preventing the action of cytokines, such as transforming growth factor-β (TGF-β).
さらに、DNA合成とエンドセリン-1を阻害し、デカペンタプレジック(Smad)シグナル伝達と細胞外シグナル調節キナーゼ1/2(ERK1/2)経路を阻害することで、線維芽細胞のコラーゲン合成と沈着を防ぎ、その結果、形質転換成長因子β(TGF-β)などのサイトカインの作用を防ぐことができます。
AcSDKP has antifibrotic and anti-inflammatory effects in the lung, heart, liver, and kidney.
AcSDKPは肺、心臓、肝臓、腎臓において抗線維化作用と抗炎症作用を有する。
Therefore, ACE may exert these effects by decreasing angiotensin II levels and increasing AcSDKP levels.
したがって、ACE はアンジオテンシン II レベルを低下させ、AcSDKP レベルを上昇させることでこれらの効果を発揮する可能性がある。
Key points:
ACE cleaves a multitude of peptides acting subcellularly, anchored to the plasma membrane, or extracellularly.
ACE は、細胞内、細胞膜に固定された、または細胞外で作用する多数のペプチドを切断します。
ACE is involved in cleaving peptides to be presented by antigen-presenting cells at MHCs. Its dysregulation may contribute to viral evasion mechanisms.
ACE は、抗原提示細胞によって MHC に提示されるペプチドの切断に関与しています。その調節異常は、ウイルス回避メカニズムに寄与する可能性があります。
ACE is overexpressed in myeloid cells as a mechanism of immune response and inflammation.
ACE は、免疫応答と炎症のメカニズムとして骨髄細胞で過剰発現しています。
ACE regulates the contact system and the KKS.
ACE は、接触システムと KKS を制御します。
ACE regulates immunity through the clearance of peptides, such as Ac-SDKP.
ACE は、Ac-SDKP などのペプチドのクリアランスを通じて免疫を制御します。
3. The Protease and Anti-Protease System in the Lung and SARS-CoV-2 Infection
肺のプロテアーゼと抗プロテアーゼシステムとSARS-CoV-2感染
The respiratory epithelium is mucosal tissue that consists of more than 40 different cell types.
呼吸器上皮は、40 種類以上の異なる細胞型からなる粘膜組織です。
These cells coordinate their actions through the release of various mediators (mucins, cytokines, proteases, and anti-proteases) to maintain respiratory function and protect against foreign agents.
これらの細胞は、さまざまなメディエーター(ムチン、サイトカイン、プロテアーゼ、抗プロテアーゼ)を放出することで、呼吸機能を維持し、外来物質から保護するために、その働きを調整します。
Proteases that are released from the respiratory mucosa are involved in processes related to airway function, including mucus characteristics (i.e., its density or rheology), mucociliary clearance, and the recruitment and function of immune cells.
呼吸器粘膜から放出されるプロテアーゼは、粘液特性(密度またはレオロジー)、粘膜繊毛クリアランス、免疫細胞の動員と機能など、気道機能に関連するプロセスに関与しています。
This process is carried out by cleaving pre-proteins to their physiologically active forms, a process that is finely regulated by the action of anti-proteases to control excessive over-activation.
このプロセスは、プレタンパク質を生理学的に活性な形態に切断することによって行われ、このプロセスは異常な過剰活性化を制御するために抗プロテアーゼの作用によって細かく制御されます。
The human genome contains more than 600 proteases, which gives an idea of their importance.
ヒトゲノムには600種類以上のプロテアーゼが含まれており、その重要性がうかがえます。
When infection occurs, the respiratory epithelium starts to produce proteases, such as the type II transmembrane serine proteases (TTSPs).
感染が起こると、呼吸器上皮はII型膜貫通セリンプロテアーゼ(TTSP)などのプロテアーゼを産生し始めます。
This family includes matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs), cathepsins, proteinase-3, human neutrophil elastase, trypsin, chymotrypsin, prostasin, and TMPRSSs.
このファミリーには、マトリックスメタロプロテアーゼ(MMP)、ディスインテグリンおよびメタロプロテアーゼ(ADAM)、トロンボスポンジンモチーフを有するディスインテグリンおよびメタロプロテアーゼ(ADAMTS)、カテプシン、プロテイナーゼ-3、ヒト好中球エラスターゼ、トリプシン、キモトリプシン、プロスタシン、およびTMPRSSが含まれます。
In addition to the aforementioned processes, these proteases regulate tissue repair, blood coagulation (secondary haemostasis), fibrinolysis, and immune function.
前述のプロセスに加えて、これらのプロテアーゼは、組織修復、血液凝固(二次止血)、線維素溶解、および免疫機能を調節します。
Moreover, anti-proteases or serpins can be released from respiratory epithelia, including α-1-antitrypsin (serpin A1), plasminogen activator inhibitor 1 (serpin E1), and glia-derived nexin (serpin E2), which can reduce SARS-CoV-2 infection.
さらに、呼吸器上皮からは、α-1-アンチトリプシン(セルピンA1)、プラスミノーゲン活性化因子阻害因子1(セルピンE1)、グリア由来ネキシン(セルピンE2)などの抗プロテアーゼまたはセルピンが放出され、SARS-CoV-2感染を軽減することができます。
The underlying pulmonary pathological processes involve an imbalance between proteases and anti-proteases, and viruses are known to deregulate this balance.
肺の病理学的プロセスの基礎には、プロテアーゼと抗プロテアーゼの不均衡が関与しており、ウイルスはこのバランスを乱すことが知られています。
Proteomics studies in cells infected in vitro with SARS-CoV-2 have shown dysregulation with increased expression of proteases and decreased expression of anti-proteases (e.g., SPINT1 (Kunitz-type protease inhibitor 1), SPINT2 (Kunitz-type protease inhibitor 2), tissue inhibitors of metalloproteinases 1/2 (TIMP1/2), amyloid beta precursor-like protein 2 (APLP2), cystatin C (CST3), and α-1-antitrypsin).
SARS-CoV-2にin vitroで感染した細胞におけるプロテオミクス研究では、プロテアーゼの発現増加と抗プロテアーゼの発現減少を伴う調節不全が示されています(例:SPINT1(クニッツ型プロテアーゼ阻害剤1)、SPINT2(クニッツ型プロテアーゼ阻害剤2)、組織メタロプロテアーゼ阻害剤1/2(TIMP1/2)、アミロイドβ前駆体様タンパク質2(APLP2)、シスタチンC(CST3)、α-1アンチトリプシン)。
Therefore, it is necessary to re-establish their balance to have a healthy mucosa.
したがって、健康な粘膜を保つには、それらのバランスを再構築する必要があります。
Key points:
The respiratory mucosa controls its functions through a complex release system of proteases and anti-proteases.
呼吸器粘膜は、プロテアーゼと抗プロテアーゼの複雑な放出システムを通じてその機能を制御します。
Respiratory viruses in their infective process unbalance the action of proteases and anti-proteases.
呼吸器ウイルスは感染過程でプロテアーゼと抗プロテアーゼの作用のバランスを崩します。
3.1. Host Proteases in SARS-CoV-2 Infection
SARS-CoV-2感染における宿主プロテアーゼ
Coronaviruses, such as SARS-CoV-2, take advantage of the activity of multiple proteases to infect a host cell.
SARS-CoV-2などのコロナウイルスは、複数のプロテアーゼの活性を利用して宿主細胞に感染します。
When respiratory epithelial cells become infected, hyperreactivity occurs, where proteases (e.g., trypsin, cathepsins, and/or elastases) are overexpressed and released from respiratory tract epithelial cells as well as myeloid cells.
呼吸器上皮細胞が感染すると、過剰反応が起こり、プロテアーゼ(トリプシン、カテプシン、エラスターゼなど)が過剰発現し、呼吸器上皮細胞および骨髄細胞から放出されます。
Excessive activity of these proteases significantly contributes to the inflammatory and/or infectious processes.
これらのプロテアーゼの過剰な活性は、炎症および/または感染プロセスに大きく寄与します。
We have already discussed the role of TTSPs, such as TMPRSS2, in the process of SARS-CoV-2 entry into a host epithelial cell.
SARS-CoV-2が宿主上皮細胞に侵入するプロセスにおけるTMPRSS2などのTTSPの役割についてはすでに説明しました。
Below, we briefly review other proteases that also play an important role in the pathological process in the respiratory tract.
以下では、呼吸器系の病理学的プロセスにおいて重要な役割を果たす他のプロテアーゼについても簡単に説明します。
3.1.1. ADAMs
ADAMs are a family of type I transmembrane proteins belonging to the adamalysin subfamily of metalloproteinases.
ADAM は、メタロプロテアーゼのアダマリシン サブファミリーに属する I 型膜貫通タンパク質のファミリーです。
The members of this family have a metalloprotease domain and an integrin-interacting domain (disintegrin domain), indicating that they have both protease and adhesion molecule activity.
このファミリーのメンバーは、メタロプロテアーゼ ドメインとインテグリン相互作用ドメイン (ディスインテグリン ドメイン) を持ち、プロテアーゼと接着分子の両方の活性があることを示しています。
They participate in the cellular processes of migration, adhesion, and cell fusion.
これらは、細胞の移動、接着、細胞融合のプロセスに関与します。
In addition, through their protease activity, they participate in cell signalling by cleaving certain protein domains (sheddase activity), leading to the release of cell membrane-associated proteins, such as cytokines, apoptotic ligands, growth factors, and receptors.
さらに、プロテアーゼ活性を通じて、特定のタンパク質ドメインを切断して (シェダーゼ活性)、細胞シグナル伝達に関与し、サイトカイン、アポトーシス リガンド、成長因子、受容体などの細胞膜関連タンパク質を放出します。
One of the most studied examples is the production of TNF-α.
最も研究されている例の 1 つは、TNF-α の生成です。
ADAM17 is also known as the TNF-α convertase (TACE)—it cleaves transmembrane TNF (26 kDa) to release its active soluble form, TNF-α (17 kDa).
ADAM17 は TNF-α 変換酵素 (TACE) としても知られており、膜貫通 TNF (26 kDa) を切断して、活性可溶性形態の TNF-α (17 kDa) を放出します。
This family of proteases also produce mucus at the bronchial level.
このプロテアーゼファミリーは気管支レベルでも粘液を生成します。
In COVID-19, ADAM17 is upregulated via internalisation of ACE2 after it binds to SARS-CoV-2, a phenomenon that has been observed with other coronaviruses.
COVID-19では、ADAM17はSARS-CoV-2に結合した後、ACE2の内在化を介してアップレギュレーションされますが、この現象は他のコロナウイルスでも観察されています。
In addition, ADAM17 proteolytically cleaves ACE2, releasing it in its soluble form (sACE2) and decreasing its expression in the cytoplasmic membrane.
さらに、ADAM17はACE2をタンパク質分解的に切断し、可溶性形態(sACE2)で放出し、細胞膜での発現を減少させます。
This phenomenon contributes to the infectious process as well as the release of TNF-α, interleukin 6 (IL-6), and other proinflammatory molecules, which aggravates the inflammatory process (Figure 1B).
この現象は感染プロセスに寄与するだけでなく、TNF-α、インターロイキン6(IL-6)、その他の炎症誘発性分子の放出にも寄与し、炎症プロセスを悪化させます(図1B)。
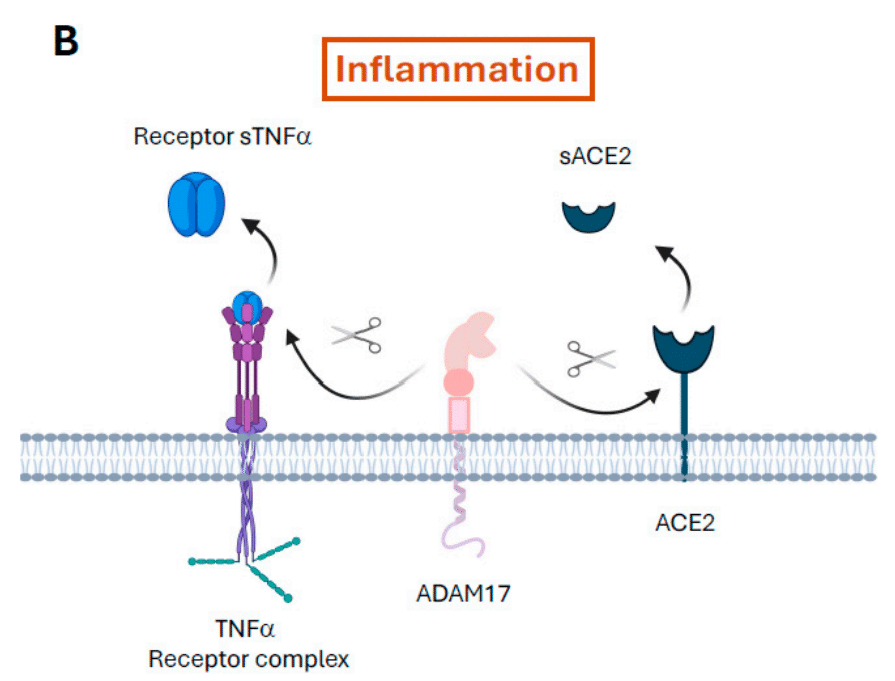
3.1.2. Elastases and Other Neutrophil Serine Proteases
エラスターゼとその他の好中球セリンプロテアーゼ
Elastases are serine proteases that have important physiological functions through the cleavage of multiple protein substrates.
エラスターゼは、複数のタンパク質基質の切断を通じて重要な生理学的機能を果たすセリンプロテアーゼです。
In coronavirus infections, elastases, such as pancreatic elastase, are involved in cleavage of the S protein via an elastase-specific domain in the S2 subunit.
コロナウイルス感染症では、膵臓エラスターゼなどのエラスターゼが、S2サブユニットのエラスターゼ特異的ドメインを介してSタンパク質の切断に関与している。
For SARS-CoV-2, the involvement of elastases released from myeloid cells such as macrophages, but mostly neutrophils, has been investigated.
SARS-CoV-2については、マクロファージなどの骨髄細胞(主に好中球)から放出されるエラスターゼの関与が調査されている。
Their importance lies in the viral entry process.
その重要性はウイルスの侵入プロセスにある。
In addition, the release of human neutrophil elastase from neutrophils is associated with increased production of cathepsins and matrix metalloproteases, which promote infection.
さらに、好中球からのヒト好中球エラスターゼの放出は、感染を促進するカテプシンおよびマトリックスメタロプロテアーゼの産生増加と関連している。
Along with human neutrophil elastase, neutrophils release other serine proteases, such as cathepsin G, proteinase-3, neutrophil serine protease-4 (NSP-4), azurocidin (AZU1), myeloperoxidase, myeloblastin (PRTN3), and transcobalamin-1 (TCN1), which are stored in azurophil granules.
ヒト好中球エラスターゼとともに、好中球はカテプシンG、プロテイナーゼ-3、好中球セリンプロテアーゼ-4(NSP-4)、アズロシジン(AZU1)、ミエロペルオキシダーゼ、ミエロブラスチン(PRTN3)、トランスコバラミン-1(TCN1)などの他のセリンプロテアーゼを放出し、これらはアズロフィル顆粒に蓄えられている。
The main functions are to degrade the extracellular matrix by digesting collagen, transmembrane proteins, pulmonary surfactant factors, and proteoglycans.
主な機能は、コラーゲン、膜貫通タンパク質、肺サーファクタント因子、プロテオグリカンを消化して細胞外マトリックスを分解することです。
Therefore, their release contributes to a process of permeabilisation of cellular barriers, inflammation, and alteration of lung functions.
したがって、それらの放出は、細胞バリアの透過性、炎症、肺機能の変化のプロセスに寄与します。
In addition, they participate in inflammation by processing and modifying cytokine functions, inhibiting anti-inflammatory factors, such as progranulin, activating surface receptors, such as Toll-like receptors (TLRs) through their protease action, and promoting cytokine release from monocytes and macrophages.
さらに、サイトカイン機能の処理と修正、プログラニュリンなどの抗炎症因子の阻害、プロテアーゼ作用によるToll様受容体(TLR)などの表面受容体の活性化、単球とマクロファージからのサイトカイン放出の促進などにより炎症に関与する。
However, in COVID-19, the most prominent role of human neutrophil elastase is in NET formation and NETosis, a process in which neutrophils expel their citrullinated chromatin into the extracellular milieu.
しかし、COVID-19では、ヒト好中球エラスターゼの最も顕著な役割は、NET形成とNETosis、つまり好中球がシトルリン化されたクロマチンを細胞外環境に排出するプロセスにあります。
Combined with the action of released proteases, a network is produced with the capacity to trap platelets and red blood cells, linking the thromboembolic processes that occur during COVID-19.
放出されたプロテアーゼの作用と相まって、血小板と赤血球を捕捉する能力を持つネットワークが生成され、COVID-19中に発生する血栓塞栓プロセスを結び付けます。
The exacerbation of these processes is partially caused by an imbalance between the release and activity of proteases secreted by neutrophils, as opposed to anti-proteases, such as α-1-antitrypsin and other inhibitors secreted from leucocytes.
これらのプロセスの悪化は、好中球から分泌されるプロテアーゼの放出と活性の不均衡と、白血球から分泌されるα-1-アンチトリプシンやその他の阻害剤などの抗プロテアーゼの放出と活性の不均衡によって部分的に引き起こされる。
3.1.3. Trypsin and Human Airway Trypsin-like Proteases (HATs)
トリプシンとヒト気道トリプシン様プロテアーゼ(HAT)
Trypsin has been shown to increase the infectivity of several coronaviruses, including SARS-CoV-2, when the virus is already attached to its cellular target.
トリプシンは、SARS-CoV-2を含むいくつかのコロナウイルスの感染力を、ウイルスがすでに細胞標的に付着している場合に高めることが示されている。
These mechanisms do not necessarily require binding to ACE2, but they favour increased membrane fusion.
これらのメカニズムは必ずしもACE2への結合を必要としないが、膜融合の増加に有利である。
In certain SARS-CoV-2 variants (e.g., Delta) that have a greater capacity to produce syncytia, and therefore have greater membrane fusion activity, trypsin plays a key role in this infectious mechanism.
シンシチウムを生成する能力が高く、したがって膜融合活性が高い特定のSARS-CoV-2変異体(デルタなど)では、トリプシンがこの感染メカニズムで重要な役割を果たしている。
On the other hand, excess trypsin release contributes to cell barrier permeabilisation, inflammation, and impaired lung function.
一方、過剰なトリプシン放出は、細胞バリアの透過性、炎症、肺機能障害に寄与する。
HATs are preferentially anchored to the surface of bronchial and tracheal respiratory tract hair cells.
HATは、気管支および気管気道の有毛細胞の表面に優先的に固定される。
They can also be found in soluble forms in patients with respiratory diseases.
呼吸器疾患の患者では可溶性形態でも見つかることがある。
Coronaviruses that need to cleave proteins from their capsid during the infectious process can use these proteases.
感染過程でカプシドからタンパク質を切断する必要があるコロナウイルスは、これらのプロテアーゼを利用できます。
In addition, they participate in the activation of epithelial sodium channels (ENaC) that hydrate the airway and facilitate mucociliary clearance.
さらに、それらは気道に水分を補給し、粘液繊毛のクリアランスを促進する上皮ナトリウムチャネル(ENaC)の活性化にも関与している。
ENaC and the coronavirus S protein share the same furin-like cleavage domain, and thus use the same proteases (e.g., TMPRSS2) for protein activation.
ENaCとコロナウイルスSタンパク質は同じフーリン様切断ドメインを共有しており、したがってタンパク質活性化に同じプロテアーゼ(例:TMPRSS2)を使用します。
Hence, by modifying the activity of proteases after infection, coronaviruses may affect ENaC activity.
したがって、感染後にプロテアーゼの活性を変化させることで、コロナウイルスはENaC活性に影響を与える可能性がある。
SARS-CoV-2 leads to the overexpression of proteases, such as TMPRSS2, at the host plasma membrane and simultaneously prevents these proteases from degrading ENaC, leaving them in an overactivated state.
SARS-CoV-2は、宿主細胞膜でTMPRSS2などのプロテアーゼの過剰発現を引き起こし、同時にこれらのプロテアーゼによるENaCの分解を妨げ、過剰活性化状態になります。
This state accelerates viral entry into the host cell.
この状態は、ウイルスの宿主細胞への侵入を加速します。
In addition, altered ENaC activity is consistent with symptoms following SARS-CoV-2 infection, such as a runny nose, ageusia, pulmonary oedema, and respiratory distress.
さらに、変化したENaC活性は、鼻水、味覚異常、肺水腫、呼吸困難などのSARS-CoV-2感染後の症状と一致しています。
The increased release or expression of proteases, such as trypsin, HATs, and others secreted by neutrophils (e.g., elastase and proteinase-3), favours activation of protease-activated receptor type 2.
トリプシン、HAT、好中球から分泌されるその他のプロテアーゼ(エラスターゼ、プロテイナーゼ-3など)などの放出または発現の増加は、プロテアーゼ活性化受容体2型の活性化を促進する。
This receptor promotes the release of inflammatory mediators that aggravate the inflammatory process and cell growth, contributing to airway remodelling through fibroblast proliferation.
この受容体は炎症過程と細胞増殖を悪化させる炎症メディエーターの放出を促進し、線維芽細胞の増殖を通じて気道リモデリングに寄与する。
In addition, they can cleave and activate the urokinase-type plasminogen-activated receptor, which also contributes to thromboembolic processes in patients with COVID-19.
さらに、ウロキナーゼ型プラスミノーゲン活性化受容体を切断して活性化することができ、これもCOVID-19患者の血栓塞栓プロセスに寄与する。
3.1.4. Cathepsins
カテプシン
Cysteine cathepsins are a family of papain-like proteases found intracellularly in organelles such as lysosomes, although they have also been observed in the cytosol, mitochondria, nucleus, plasma membranes, and the extracellular milieu.
システインカテプシンは、リソソームなどの細胞内器官に存在するパパイン様プロテアーゼのファミリーですが、細胞質、ミトコンドリア、核、細胞膜、細胞外環境でも観察されています。
Cathepsin secretion into the extracellular milieu is observed under physiological conditions; for example, cathepsin B, K, and L are secreted from thyroid epithelial cells to release thyroid hormones from thyroglobulins.
カテプシンの細胞外への分泌は生理的条件下で観察されます;たとえば、カテプシン B、K、L は甲状腺上皮細胞から分泌され、甲状腺グロブリンから甲状腺ホルモンを放出します。
However, excessive cathepsin release can also be triggered in pathological conditions that involve inflammation.
ただし、炎症を伴う病的な状態では、カテプシンの過剰放出が引き起こされることもあります。
In infectious diseases, such as COVID-19, where inflammation is evident, their expression and secretion increase.
COVID-19などの炎症が明らかな感染症では、それらの発現と分泌が増加します。
A relevant fact is the ability of cathepsins to modify the spatial conformation of proteins by cleaving certain domains to alter their functionality.
関連する事実として、カテプシンは特定のドメインを切断してタンパク質の空間的構造を変更し、その機能を変化させる能力がある。
The high transmissibility of SARS-CoV-2 variants, such as Omicron, has been linked to a greater capacity to enter the cell via an endocytic pathway, thanks to cathepsin-mediated modification of the S protein to produce the correct spatial arrangement of the S2 subunit.
オミクロンなどのSARS-CoV-2変異体の高い伝染性は、Sタンパク質のカテプシン媒介修飾によりS2サブユニットの正しい空間配置が生成され、エンドサイトーシス経路を介して細胞に侵入する能力が高くなったことと関連している。
A large number of cathepsin subtypes have been proposed to be involved in SARS-CoV-2 infection.
SARS-CoV-2感染には、多数のカテプシンサブタイプが関与していると提案されている。
Researchers have used in silico and in vitro approaches to determine the exact S protein cleavage sites.
研究者は、S タンパク質の正確な切断部位を決定するために、コンピューターによる手法と試験管内手法を用いてきた。
Cathepsins are upregulated upon infection, which contributes to aggravating infection, especially in those tissues where other proteases, such as TMPRSS2, are not as present, thus allowing a new entry route to infect these cells.
カテプシンは感染時にアップレギュレーションされ、特にTMPRSS2などの他のプロテアーゼが存在しない組織では感染を悪化させる一因となり、これらの細胞に感染する新しい侵入経路を可能にします。
Another relevant factor regarding the action of cathepsins in the extracellular milieu is that they are important initiators or suppressors of the activity of other proteases or cytogens involved in proteolytic cascades.
細胞外環境におけるカテプシンの作用に関するもう 1 つの関連要因は、カテプシンがタンパク質分解カスケードに関与する他のプロテアーゼまたは細胞原の活性の重要な開始因子または抑制因子であることです。
For example, cathepsin G is one of the main proteases, along with human neutrophil elastase, involved in NET formation, inflammation, and thrombosis.
たとえば、カテプシン G は、ヒト好中球エラスターゼとともに、NET 形成、炎症、および血栓症に関与する主要なプロテアーゼの 1 つです。
The mechanisms involve inactivation of tissue factor inhibitory peptide and activation of protease-activated receptors.
そのメカニズムには、組織因子阻害ペプチドの不活性化とプロテアーゼ活性化受容体の活性化が含まれます。
In addition, cathepsins are potent activators of blood platelets, and cathepsin L activates the KKS.
さらに、カテプシンは血小板の強力な活性化因子であり、カテプシンLはKKSを活性化します。
Cathepsin F, L, S, and V are also present in endolysosomes, where they cleave proteins and contribute to maturation and processing functions.
カテプシンF、L、S、Vはエンドリソソームにも存在し、そこでタンパク質を切断し、成熟と処理機能に寄与する。
An example of these functions is the processing of ENaC subunits.
これらの機能の一例は、ENaCサブユニットの処理である。
The presence of SARS-CoV-2 and its viral proteins in lysosomes interferes in the proteolytic cleavage of ENaC and in its maturation processes that alter its functionality.
SARS-CoV-2とそのウイルスタンパク質がリソソームに存在すると、ENaCのタンパク質分解による切断と、その機能を変化させる成熟プロセスが妨げられる。
These changes contribute to COVID-19 symptoms, as ENaC is involved in proper homeostasis of the pulmonary fluid interface and in the occurrence of electrolyte imbalance (i.e., hypokalaemia) due to altered renal function.
これらの変化はCOVID-19の症状に寄与しており、ENaCは肺液界面の適切な恒常性と、腎機能の変化による電解質不均衡(すなわち低カリウム血症)の発生に関与している。
Another example is the processing of antigenic proteins in immune cells for presentation by MHC-I and MHC-II.
もう 1 つの例は、MHC-I および MHC-II による提示のために免疫細胞で抗原タンパク質を処理することです。
Moreover, lysosomal cathepsins influence the trafficking of SARS-CoV-2-specific proteins, such as the accessory protein open reading frame 3a (ORF3a), which is involved in virus infectivity and the formation of new virions.
さらに、リソソームカテプシンは、ウイルスの感染性と新しいウイルス粒子の形成に関与するアクセサリータンパク質オープンリーディングフレーム3a(ORF3a)などのSARS-CoV-2特異的タンパク質の輸送に影響を与えます。
Indeed, if cathepsins are inhibited, it can lead to degradation of new virions into cellular multivesicular bodies.
実際、カテプシンが阻害されると、新しいウイルス粒子が細胞多小胞体に分解される可能性がある。
SARS-CoV-2 may use a lysosomal pathway that involves cathepsins to release newly formed virions from the host cell, although the mechanisms remain unclear.
SARS-CoV-2は、カテプシンが関与するリソソーム経路を使用して、新たに形成されたウイルス粒子を宿主細胞から放出する可能性があるが、そのメカニズムは不明である。
In addition, cathepsin L is involved in the upregulation and processing of heparanase, which is implicated in the release of viral progeny and their propagation.
さらに、カテプシンLはヘパラナーゼのアップレギュレーションと処理に関与しており、ウイルスの子孫の放出と増殖に関与している。
Serum heparanase levels are increased in patients with COVID-19 and correlate with the severity of the disease.
COVID-19 患者では血清ヘパラナーゼ濃度が上昇しており、病気の重症度と相関している。
Specifically, heparanase damages the glycocalyx of endothelial cells and promotes thromboembolism.
具体的には、ヘパラナーゼは内皮細胞のグリコカリックスを損傷し、血栓塞栓症を促進します。
It causes the release of certain molecules from heparan sulphate proteoglycans of the glycocalyx that bind to it, including growth factors, cytokines, enzymes, and lipoproteins.
カテプシンは、グリコカリックスのヘパラン硫酸プロテオグリカンから特定の分子を放出し、成長因子、サイトカイン、酵素、リポタンパク質などが含まれます。
These molecules released by heparanase are involved in cell motility, angiogenesis, inflammation, coagulation, stimulation of autophagy, and exosome production.
ヘパラナーゼによって放出されるこれらの分子は、細胞運動、血管新生、炎症、凝固、オートファジーの刺激、およびエクソソーム生成に関与しています。
Since cathepsins are involved in the mechanisms of entry, processing of viral proteins, and release of viral progeny to the cellular exterior, their inhibition is a very attractive and well-studied therapeutic strategy for the development of antiviral drugs against coronaviruses.
カテプシンは、ウイルスタンパク質の侵入、処理、および細胞外へのウイルス子孫の放出のメカニズムに関与しているため、その阻害はコロナウイルスに対する抗ウイルス薬の開発のための非常に魅力的でよく研究された治療戦略です。
Key points:
SARS-CoV-2 infection upregulates ADAM-17, causing the adhesion of inflammatory cells and the release of chemoattractants, such as sACE2 and proinflammatory cytokines (IL-6 and TNF-α).
SARS-CoV-2 感染により ADAM-17 がアップレギュレーションされ、炎症細胞の接着と、sACE2 や炎症性サイトカイン (IL-6 および TNF-α) などの化学誘引物質の放出が引き起こされます。
Elastases participate in viral entry into the host cell and the release of other proteases that amplify the infectious and inflammatory response, permeabilise cellular barriers, and alter lung functions. Neutrophil elastase participates in NETosis.
エラスターゼは、ウイルスの宿主細胞への侵入と、感染および炎症反応を増幅し、細胞バリアを透過性にし、肺機能を変化させる他のプロテアーゼの放出に関与します。好中球エラスターゼは NETosis に関与します。
Trypsin promotes infection when the virus is attached to the cell membrane by increasing the membrane fusion process.
トリプシンは、ウイルスが細胞膜に付着すると、膜融合プロセスを増加させて感染を促進します。
Coronaviruses prevent proteases from exercising their physiological functions of correct maturation of proteins, such as ENaC. This is involved in many of the symptoms caused by respiratory viruses.
コロナウイルスは、ENaC などのタンパク質の適切な成熟というプロテアーゼの生理学的機能の発揮を妨げます。これは、呼吸器ウイルスによって引き起こされる多くの症状に関係しています。
Cathepsins allow the entry of SARS-CoV-2 via the endocytic route into those cells where other proteases, such as TMPRSS2, are not so present.
カテプシンは、TMPRSS2 などの他のプロテアーゼがあまり存在しない細胞に、エンドサイトーシス経路を介して SARS-CoV-2 が侵入することを可能にします。
Cathepsins initiate and amplify the activation of pathways activated by proteases such as KKS, coagulation, or the formation of NETs.
カテプシンは、KKS、凝固、または NET の形成などのプロテアーゼによって活性化される経路の活性化を開始および増幅します。
Cathepsins participate in entry, maturation of viral proteins, and release of new viral progeny from the host cell.
カテプシンは、侵入、ウイルスタンパク質の成熟、および宿主細胞からの新しいウイルス子孫の放出に関与します。
3.2. Host Anti-Proteases in SARS-CoV-2 Infection
SARS-CoV-2感染における宿主抗プロテアーゼ
3.2.1. α-1-Antitrypsin (Serpin A1)
α-1-アンチトリプシン (セルピン A1)
In addition to increasing protease activity, coronaviruses negatively modulate the action of anti-proteases, which contributes to an imbalance between proteases and anti-proteases that prevents proper respiratory function.
コロナウイルスはプロテアーゼ活性を高めるだけでなく、抗プロテアーゼの作用を負に調節し、プロテアーゼと抗プロテアーゼの不均衡に寄与して適切な呼吸機能を妨げます。
In general, coronaviruses increase the degradation of α-1-antitrypsin (serpin A1), increasing its degradation products.
一般的に、コロナウイルスはα-1-アンチトリプシン(セルピンA1)の分解を促進し、その分解産物を増加させます。
This mechanism is associated with increased pathogenicity because α-1-anthrypsin is one of the most important inhibitors of serine proteases in the lung and, therefore, has important anti-inflammatory actions.
このメカニズムは、α-1-アントリプシンが肺のセリンプロテアーゼの最も重要な阻害剤の1つであり、したがって重要な抗炎症作用があるため、病原性の増加に関連しています。
The loss of α-1-anthrypsin activity is also relevant to the SARS-CoV-2 infectious process.
α-1-アントリプシン活性の喪失は、SARS-CoV-2の感染プロセスにも関連している。
For example, α-1-antitrypsin inhibits TMPRSS2, which is required for S protein cleavage, thus aggravating the infection.
例えば、α-1-アンチトリプシンは、Sタンパク質の切断に必要なTMPRSS2を阻害します。 そのため、感染が悪化します。
Once infection has occurred, the inflammatory process starts, with IL-6 serving as one of the main mediators and a prognostic marker of the disease.
感染が発生すると炎症プロセスが始まり、IL-6が主要なメディエーターの1つとして機能し、病気の予後マーカーとなります。
One of the main actions of IL-6 is to induce hepatic synthesis of α-1-antitrypsin.
IL-6 の主な作用の 1 つは、肝臓での α-1-アンチトリプシンの合成を誘導することです。
It does so through the formation of IL-6-soluble IL-6 receptor (sIL-6R) complexes and subsequent binding to glycoprotein 130 (gp130), which activates the Janus kinase (JAK)/signal transducer and activator of transcription (STAT), ERK, and phosphoinositide 3-kinase (PI3K) signalling pathways in liver cells, regulating gene transcription.
これは、IL-6 可溶性 IL-6 受容体 (sIL-6R) 複合体の形成とそれに続く糖タンパク質 130 (gp130) への結合によって行われ、肝細胞内の Janus キナーゼ (JAK)/シグナル伝達および転写活性化因子 (STAT)、ERK、およびホスホイノシチド 3-キナーゼ (PI3K) シグナル伝達経路を活性化し、遺伝子転写を制御します。
As we discuss later, this is due to an increase in the release of sIL-6R via the action of proteases, such as ADAM17, as opposed to action on un-cleaved membrane receptors released by proteases.
後ほど説明するように、これはプロテアーゼによって放出される未切断膜受容体に対する作用ではなく、ADAM17などのプロテアーゼの作用によるsIL-6Rの放出の増加によるものです。
Hence, an increased IL-6/ α-1-antitrypsin ratio has been proposed as a biomarker for a poor prognosis in SARS-CoV-2 infection.
したがって、IL-6/α-1-アンチトリプシン比の増加は、SARS-CoV-2感染の予後不良のバイオマーカーとして提案されています。
The fact that anti-proteases, such as α-1-antitrypsin—whose function is to inhibit proteases (e.g., human neutrophil elastase, cathepsins, and metalloproteases) that are activated when the infectious process occurs, as a regulatory control mechanism—lose functionality results in infection and inflammation.
感染過程が発生したときに活性化されるプロテアーゼ(ヒト好中球エラスターゼ、カテプシン、メタロプロテアーゼなど)を阻害する機能を持つα-1-アンチトリプシンなどの抗プロテアーゼが機能を失うと、感染や炎症が発生します。
In addition, it is involved in the regulation of coagulation by inhibiting thrombin.
さらに、トロンビンを阻害することで凝固の調節にも関与しています。
Thus, patients with COVID-19 and attenuated α-1-antitrypsin production tend to have a poor prognosis.
そのため、COVID-19に罹患し、α-1-アンチトリプシン産生が低下している患者は予後が悪い傾向がある。
Therefore, α-1-antitrypsin is being evaluated for its therapeutic utility in several trials for the treatment of COVID-19 (ClinicalTrials.gov IDs: NCT04385836, NCT04547140, NCT04495101, and NCT04817332).
そのため、α-1-アンチトリプシンは、COVID-19の治療を目的としたいくつかの試験でその治療的有用性が評価されています。
3.2.2. CST3
Cystatins are a superfamily of proteins that are ubiquitously expressed in all nucleated cells and consist of at least one 100–120 amino acid inhibitory domain with protease activity.
シスタチンは、すべての核細胞で普遍的に発現するタンパク質のスーパーファミリーであり、プロテアーゼ活性を持つ少なくとも1つの100〜120アミノ酸阻害ドメインで構成されています。
There are three types of cystatins: type I or Stefins, which are cytosolic proteins, type II, which are secreted from cells into the extracellular milieu, and type III or quininogens, which are multifunctional proteins found in the blood and other fluids.
シスタチンには3つのタイプがあります。タイプIまたはステフィンは細胞質タンパク質、タイプIIは細胞から細胞外環境に分泌され、タイプIIIまたはキニノーゲンは血液やその他の体液に含まれる多機能タンパク質です。
CST3 is secreted in body fluids, such as saliva and urine.
CST3 は唾液や尿などの体液中に分泌されます。
It has antiviral activity against coronaviruses and, therefore, its recombinant forms have been proposed as antiviral drugs against them.
シスタチンはコロナウイルスに対して抗ウイルス活性があり、そのため、その組み換え型はコロナウイルスに対する抗ウイルス薬として提案されている。
The antiviral mechanism is related to the ability to inhibit cysteine proteases, such as cathepsins, which are used by the virus to gain entry to a host cell via the endocytic pathway.
抗ウイルス機構は、ウイルスがエンドサイトーシス経路を介して宿主細胞に侵入するために使用するカテプシンなどのシステインプロテアーゼを阻害する能力に関連しています。
Cathepsin S and L, among others, have an important role in foreign protein digestion, processing, and loading onto MHC-I and MHC-II for presentation by antigen-presenting cells (including dendritic cells) to T lymphocytes.
カテプシンSとLは、外来タンパク質の消化、処理、および抗原提示細胞(樹状細胞を含む)によるTリンパ球への提示のためのMHC-IおよびMHC-IIへのロードにおいて重要な役割を果たしている。
By inhibiting cathepsins, cystatins regulate the generation of peptidergic MHC antigenic complexes.
カテプシンを阻害することにより、シスタチンはペプチド性MHC抗原複合体の生成を調節する。
This process involves other related proteins that have the capacity to inhibit cathepsins.
このプロセスには、カテプシンを阻害する能力を持つ他の関連タンパク質が関与している。
The p41 isoform of CD74 of MHC-II has recently been found to inhibit them, in addition to its antigen-presenting function.
MHC-IIのCD74のp41アイソフォームは、抗原提示機能に加えて、カテプシンを阻害することが最近判明した。
In turn, CD74 can be activated through the MHC-II master regulator MHC-II transactivator (CIITA).
次に、CD74はMHC-IIマスターレギュレーターMHC-IIトランスアクチベーター(CIITA)を介して活性化される可能性がある。
Interestingly, CIITA levels have been observed to decrease in both children and adults when the prognosis of COVID-19 is severe.
興味深いことに、COVID-19の予後が重篤な場合、小児と成人の両方でCIITAレベルが低下することが観察されています。
The importance of this chain of events—ranging from CIITA activation of p41 of CD74 to cathepsins as final effector proteins—is that in addition to participating in antigen presentation, they intervene in the antiviral response via interferon (IFN).
この一連のイベント(CD74のp41のCIITA活性化から最終エフェクタータンパク質としてのカテプシンまで)の重要性は、抗原提示に参加することに加えて、インターフェロン(IFN)を介して抗ウイルス反応に介入することです。
Finally, inhibition of cathepsins causes virions to be redirected to a degradation pathway in multivesicular bodies or lysosomes.
最後に、カテプシンの阻害により、ウイルス粒子は多小胞体またはリソソーム内の分解経路にリダイレクトされます。
Hence, inactivation of these pathways plays a very important protective role via reduction of viral transcription, assembly, and release.
したがって、これらの経路の不活性化は、ウイルスの転写、組み立て、放出の減少を介して非常に重要な保護的役割を果たします。
Paradoxically, many studies on COVID-19 have reported a positive correlation between elevated serum and urine CST3 levels and mortality.
逆説的に、COVID-19に関する多くの研究では、血清および尿中のCST3レベルの上昇と死亡率の間に正の相関関係があることが報告されている。
CST3 is a highly sensitive biomarker in the assessment of cardiovascular and renal function because it is filtered only at the glomerular level.
CST3は糸球体レベルでのみ濾過されるため、心血管機能と腎機能の評価において非常に感度の高いバイオマーカーです。
Of note, renal failure is one of the main causes of mortality in patients with COVID-19.
注目すべきことに、腎不全はCOVID-19患者の死亡の主な原因の1つです。
On the other hand, in the saliva of patients with moderate or severe COVID-19, CST3 levels have been observed to be decrease with respect to healthy individuals.
一方、中等症または重症のCOVID-19患者の唾液では、健康な人に比べてCST3レベルが低下することが観察されています。
In hospitalised patients, CST3 levels tend to increase in those who are symptomatic relative to those who are asymptomatic, although these values are always lower than in healthy individuals.
入院患者では、無症状の患者と比較して症状のある患者の方がCST3レベルが上昇する傾向があるが、これらの値は健康な個人よりも常に低い。
These differences in CST3 saliva and serum levels are most likely due to renal impairment and the fact that many of these patients are treated with glucocorticoids that increase CST3 plasma levels.
CST3唾液と血清レベルのこれらの違いは、腎機能障害と、これらの患者の多くがCST3血漿レベルを上昇させるグルココルチコイドで治療されているという事実によるものと考えられます。
In addition, elevated levels of ILs, such as IL-6, downregulate cystatin levels, and vice versa.
さらに、IL-6などのILの上昇はシスタチンレベルを低下させ、逆もまた同様である。
Critically ill patients with COVID-19 have high IL-6 levels and show an abnormal glomerular filtration rate (GFR).
COVID-19 の重症患者は IL-6 値が高く、糸球体濾過率 (GFR) が異常です。
Specifically, the GFRcystatinC/eGFRcreatinine ratio is <0.6 due to the influence that cytokines, such as IL-6, have on the regulation of CST3, among other factors.
具体的には、IL-6 などのサイトカインが CST3 の調節に及ぼす影響などにより、GFR シスタチン C/eGFR クレアチニン比は 0.6 未満です。
3.2.3. Other Anti-Proteases
Other anti-proteases that are affected after SARS-CoV-2 infection include secretory leucocyte protease inhibitor (SLPI) and elafin.
SARS-CoV-2感染後に影響を受ける他の抗プロテアーゼには、分泌型白血球プロテアーゼ阻害剤(SLPI)とエラフィンがある。
SLPI is one of the most important anti-proteases secreted by clear and goblet epithelial cells, submucosal glands, and leucocytes in the airways as a protective mechanism against damage.
SLPIは、気道の透明上皮細胞、杯状上皮細胞、粘膜下腺、白血球から損傷に対する保護機構として分泌される最も重要な抗プロテアーゼの1つです。
In COVID-19, SLPI is released as an anti-inflammatory response after cytokine release.
COVID-19では、サイトカイン放出後の抗炎症反応としてSLPIが放出される。
Factors such as pulmonary surfactant A released from type II pneumocytes may contribute to its release, and infection of these cells by SARS-CoV-2 may impair these mechanisms.
II型肺胞上皮細胞から放出される肺サーファクタントAなどの因子が、その放出に寄与している可能性があり、これらの細胞がSARS-CoV-2に感染すると、これらのメカニズムが損なわれる可能性がある。
In patients with COVID-19, although increased expression has been observed , it is not able to attenuate the action of proteases, such as human neutrophil elastase.
COVID-19患者では、発現の増加が観察されているものの、ヒト好中球エラスターゼなどのプロテアーゼの作用を減弱させることはできない。
In COVID-19, there is an increased release of proteases, such as MMP-9 and/or elastase released from neutrophils, which may contribute to the clearance of SLPI.
COVID-19では、好中球から放出されるMMP-9やエラスターゼなどのプロテアーゼの放出が増加し、SLPIの除去に寄与している可能性があります。
In severe cases of COVID-19, this intense protease activity leads to increased oxidative stress, which promotes neutrophil activation.
COVID-19の重症例では、この強力なプロテアーゼ活性により酸化ストレスが増加し、好中球の活性化が促進されます。
This leads to the suppression of pathways, such as nuclear factor erythroid 2-related factor 2 (Nrf2), to poise this oxidative balance and, consequently, an imbalance in anti-protease activity.
これにより、核因子赤血球2関連因子2(Nrf2)などの経路が抑制され、この酸化バランスが均衡し、結果として抗プロテアーゼ活性の不均衡が生じます。
The administration of SLPI in its recombinant form has been proposed as a therapeutic alternative to treat COVID-19.
組み換え型のSLPIの投与は、COVID-19の治療の代替療法として提案されています。
Key points:
Coronaviruses increase the degradation of anti-proteases, such as α-1-antitrypsin.
コロナウイルスは、α-1-アンチトリプシンなどの抗プロテアーゼの分解を促進します。
Cystatin is one of the main anti-proteases that regulate the action of cathepsins.
シスタチンは、カテプシンの作用を調節する主要な抗プロテアーゼの 1 つです。
4. Consequences of SARS-CoV-2 Anchoring to ACE2
SARS-CoV-2がACE2に結合することによる影響
Cells of the respiratory epithelium are targets for more than 200 viruses, which infect them through recognition and anchoring to proteins on their plasma membrane.
呼吸器上皮細胞は200種類以上のウイルスの標的であり、ウイルスは細胞膜上のタンパク質を認識して固定することで感染します。
Several binding proteins for SARS-CoV-2 have been proposed, including neurophilin-1, metabotropic glutamate receptor subtype 2 (mGluR2), kidney injury molecule-1, heat shock protein A5 (HSPA5) or glucose-regulated protein-78 (GRP78), basigin (CD147), heparan sulphate, dipeptidyl peptidase 4/CD26, GRP78, CD147, and TLR4.
SARS-CoV-2の結合タンパク質としては、ニューロフィリン-1、代謝型グルタミン酸受容体サブタイプ2(mGluR2)、腎障害分子-1、熱ショックタンパク質A5(HSPA5)またはグルコース調節タンパク質-78(GRP78)、バシジン(CD147)、ヘパラン硫酸、ジペプチジルペプチダーゼ4/CD26、GRP78、CD147、TLR4などが提案されている。
However, ACE2 is considered the most important target because it is the one that can best explain the pathological events that occur after infection.
しかし、ACE2は感染後に起こる病理学的イベントを最もよく説明できるため、最も重要なターゲットと考えられています。
Its expression levels in different tissues predict the main target organs of SARS-CoV-2: digestive tract (ileum) > heart > kidney and urinary bladder > respiratory tract and lung >>> stomach and liver.
さまざまな組織でのその発現レベルから、SARS-CoV-2の主な標的臓器が予測されます。消化管(回腸)>心臓>腎臓と膀胱>呼吸器と肺>>>胃と肝臓。
Specifically, ACE2 expression is much higher in alveolar tissue (type II pneumocytes) relative to the other parts of the respiratory tract, explaining the great damage the SARS-CoV-2 infection causes there.
具体的には、ACE2の発現は、呼吸器の他の部分と比較して肺胞組織(II型肺胞上皮細胞)ではるかに高く、SARS-CoV-2感染がそこで大きな損傷を引き起こすことを説明しています。
With the entry of the virus through the airway, and the subsequent proteolytic cleavage of the S protein by host proteases, the S1 subunit interacts with ACE2 exposed on the surface of mucosal cells.
ウイルスが気道から侵入し、その後宿主プロテアーゼによってSタンパク質が分解されると、S1サブユニットは粘膜細胞の表面に露出したACE2と相互作用する。
This anchoring facilitates the entry of the virus into epithelial cells.
この固定により、ウイルスは上皮細胞に侵入しやすくなります。
The mechanism involves internalisation of ACE2 and its degradation by a protein that regulates cholesterol: protein convertase subtilisin-kexin type 9 (PCSK9).
そのメカニズムには、ACE2 の内在化と、コレステロールを制御するタンパク質、タンパク質転換酵素サブチリシン-ケキシン 9 型 (PCSK9) によるその分解が含まれます。
Therefore, there is a reduction in ACE2 expression on the surface of the membranes of infected cells.
したがって、感染細胞の膜表面でのACE2発現は減少する。
This reduction is not only due to internalisation and degradation.
この減少は、内部化と分解によるものだけではありません。
In addition, coronaviruses downregulate ACE2 mRNA levels in tissues, such as the lung and myocardium.
さらに、コロナウイルスは肺や心筋などの組織におけるACE2 mRNAレベルをダウンレギュレーションします。
ACE2 expression levels in the lung have been associated with a protective factor against respiratory distress.
肺におけるACE2の発現レベルは、呼吸困難に対する保護因子と関連している。
A partial explanation for ACE2 upregulation caused by the virus is linked to the overexpression of ADAM17 via a mechanism that is still unclear.
ウイルスによって引き起こされるACE2の上方制御の部分的な説明は、まだ不明なメカニズムを介してADAM17の過剰発現に関連しています。
ADAM17 is involved in the removal of ACE2 ectodomains from the cytoplasmic membrane, which consequently leads to an increase in free sACE2 (Figure 1B).
ADAM17は、ACE2エクトドメインを細胞膜から除去することに関与しており、その結果、遊離sACE2が増加します(図1B)。
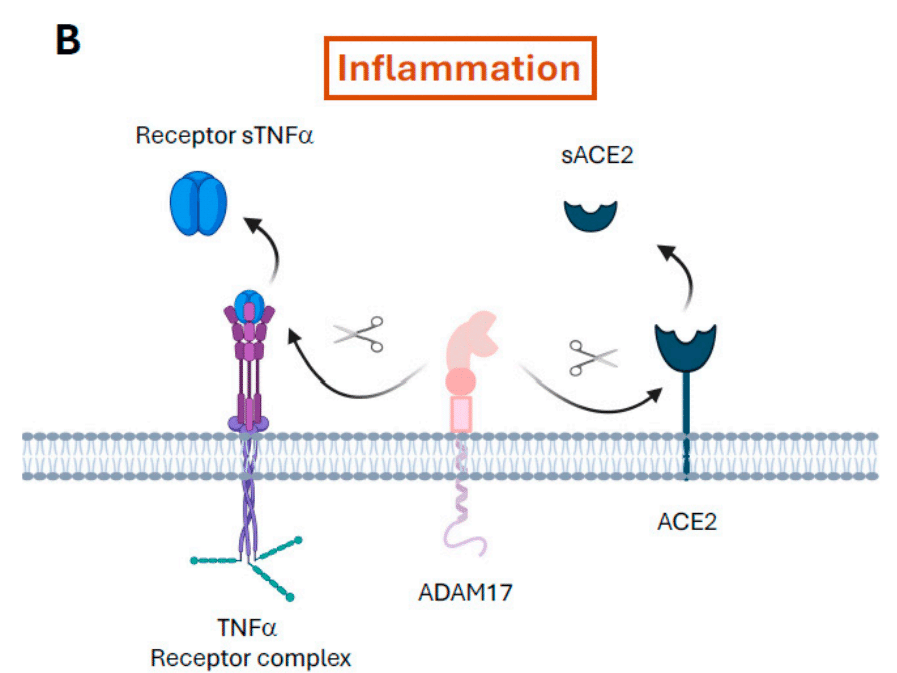
In addition, it also causes the release from the plasma membrane of proinflammatory factors, such as TNF-α and its receptors TNF receptor 1 and 2.
さらに、TNF-αやその受容体であるTNF受容体1および2などの炎症誘発因子の細胞膜からの放出も引き起こします。
The same is true for IL-6R: the release of sIL-6R and the formation of IL6–sIL-6R complexes, which in turn bind to gp130 that is expressed on many cell membranes, exacerbates IL-6 production through the JAK/STAT3 pathway.
IL-6Rについても同様です。sIL-6Rの放出とIL6-sIL-6R複合体の形成は、多くの細胞膜に発現しているgp130に結合し、JAK/STAT3経路を介してIL-6産生を悪化させます。
Autopsy studies of patients who died from coronavirus infections revealed that ACE2 cells, which are infected by the virus, produce the most proinflammatory cytokines.
コロナウイルス感染症で死亡した患者の剖検研究により、ウイルスに感染したACE2細胞が最も炎症性サイトカインを産生することが明らかになった。
Furthermore, increased accumulation of sACE2 throughout the infectious process correlates independently with mortality in SARS-CoV-2 infection.
さらに、感染過程全体にわたるsACE2の蓄積の増加は、SARS-CoV-2感染における死亡率と独立して相関している。
Thus, the accumulation of sACE2 is a measure by ADAM17 activity and is related to the inflammatory process.
したがって、sACE2 の蓄積は ADAM17 活性の尺度であり、炎症プロセスに関連しています。
On the other hand, ACE2 downregulation leads to an imbalance in the production of angiotensinogen-derived peptides and, consequently, an imbalance between the angiotensin II/angiotensin II receptor type 1 (AT1R) and its counterpart angiotensin 1–7/MAS receptor pathways.
一方、ACE2 のダウンレギュレーションは、アンジオテンシノーゲン由来ペプチドの産生の不均衡につながり、その結果、アンジオテンシン II/アンジオテンシン II 受容体タイプ 1 (AT1R) とその対応するアンジオテンシン 1-7/MAS 受容体経路間の不均衡につながります。
In addition, ACE2 is responsible for the degradation of other peptides, such as apelin (an APJ receptor agonist) and des-Arg+9-bradykinin (a bradykinin receptor B1 agonist), linking this protease to the KKS.
さらに、ACE2はアペリン(APJ受容体アゴニスト)やdes-Arg+9-ブラジキニン(ブラジキニン受容体B1アゴニスト)などの他のペプチドの分解にも関与しており、このプロテアーゼをKKSに結び付けています。
We will discuss the importance of bradykinin in COVID-19 later.
COVID-19におけるブラジキニンの重要性については後ほど説明します。
One of the main consequences of ACE2 downregulation is increased production of angiotensin II, which is closely correlated with the viral load.
ACE2のダウンレギュレーションの主な結果の1つは、ウイルス量と密接に相関するアンジオテンシンIIの産生増加です。
Angiotensin II acts on the AT1R, which has been implicated in the control of blood pressure and electrolyte balance.
アンジオテンシン II は AT1R に作用し、血圧と電解質バランスの制御に関与していると考えられています。
However, after SARS-CoV-2 infection, AT1R exacerbates vasoconstriction, inflammation, cell proliferation, fibrosis, thrombosis, and oxidative stress via ROS production.
しかし、SARS-CoV-2 感染後、AT1R は ROS 生成を介して血管収縮、炎症、細胞増殖、線維症、血栓症、酸化ストレスを悪化させます。
AT1R is expressed on myeloid cells (dendritic cells and macrophages), neutrophils, mononuclear cells, T and B lymphocytes, and non-immune tissue cells.
AT1R は、骨髄細胞(樹状細胞およびマクロファージ)、好中球、単核細胞、T リンパ球および B リンパ球、および非免疫組織細胞に発現しています。
The latter are the most reactive.
後者は最も反応性が高いです。
Their activation results in the release of inflammatory mediators, such as vascular endothelial growth factor (VEGF), prostaglandins, TNF-α, IL-1β, IL-6, IL-10, and ROS.
これらの活性化により、血管内皮増殖因子(VEGF)、プロスタグランジン、TNF-α、IL-1β、IL-6、IL-10、ROSなどの炎症メディエーターが放出されます。
These actions are mediated by multiple signalling pathways, including NOX, NF-κB, ERK1/2, MAPK, and STAT1.
これらの作用は、NOX、NF-κB、ERK1/2、MAPK、STAT1などの複数のシグナル伝達経路によって媒介されます。
These signalling pathways are the main molecular players in triggering a hyperinflammatory state (cytokine storm) and acute respiratory distress syndrome.
これらのシグナル伝達経路は、過炎症状態(サイトカインストーム)および急性呼吸窮迫症候群を引き起こす主な分子因子である。
These mechanisms provide insight into why angiotensin II leads to apoptosis of the alveolar epithelium through AT1R.
これらのメカニズムは、アンジオテンシン II が AT1R を介して肺胞上皮のアポトーシスを引き起こす理由についての洞察を提供します。
Key points:
ACE2 is the most important entry target of SARS-CoV-2 since the dysfunction in its activity caused by the infection best explains the COVID-19 disease.
ACE2 は SARS-CoV-2 の最も重要な侵入ターゲットです。感染によって引き起こされるその活動の機能不全が COVID-19 疾患を最もよく説明しているからです。
Viral entry causes an overexpression of ADAM17, a decrease in ACE2 in the plasma membrane, and an increase in its soluble forms, which causes inflammation due to the release of TNF-α and IL-6.
ウイルスの侵入により、ADAM17 の過剰発現、細胞膜における ACE2 の減少、およびその可溶性形態の増加が引き起こされ、TNF-α および IL-6 の放出による炎症を引き起こします。
Dysregulation of ACE2 causes an increase in angiotensin II that contributes to the hyperinflammatory state, respiratory distress, and damage to the lung epithelium.
ACE2 の調節不全によりアンジオテンシン II が増加し、過炎症状態、呼吸困難、および肺上皮の損傷につながります。
4.1. Dysregulation of ACE2 and Its Relation to Bradykinin
ACE2の調節異常とブラジキニンとの関係
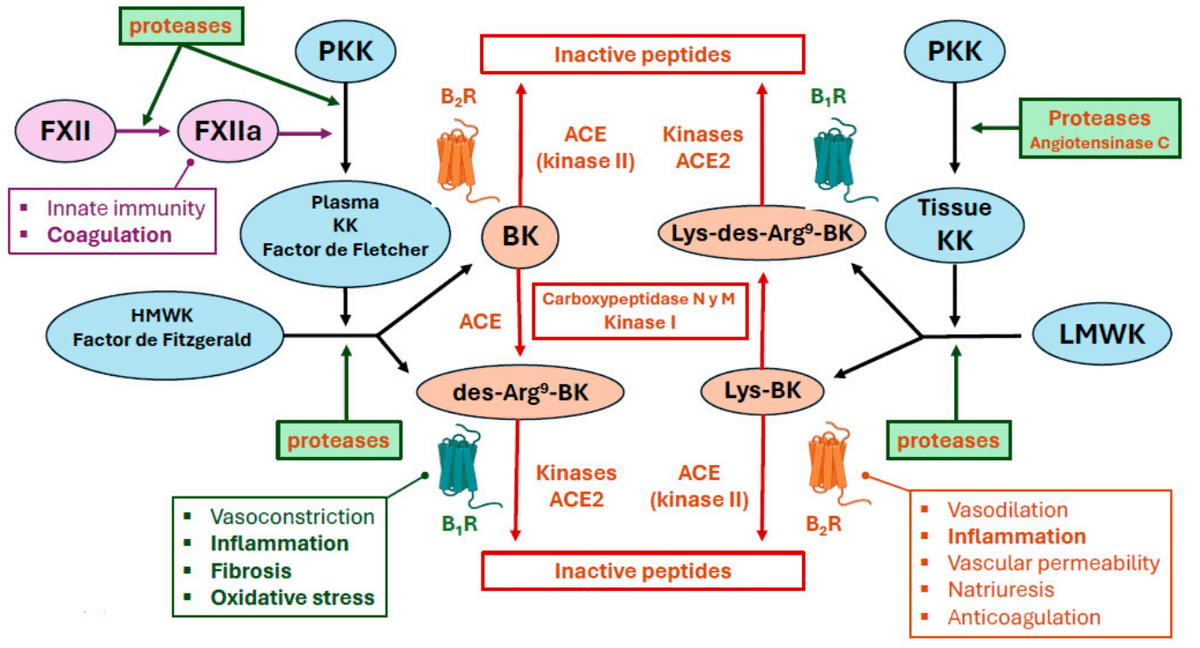
Kinins, including bradykinin, require the action of plasma or tissue kallikrein for their synthesis.
ブラジキニンを含むキニンは、その合成に血漿または組織カリクレインの作用を必要とする。
In the plasma, pre-kallikrein (Fletcher factor) is cleaved, transforming it to an enzyme with serine-protease activity.
血漿中では、プレカリクレイン(フレッチャー因子)が切断され、セリンプロテアーゼ活性を持つ酵素に変換されます。
Kallikrein cleaves a pre-protein kininogen to produce high-molecular-weight kininogen (HMWK; also known as Fitzgerald factor) and bradykinin.
カリクレインはプレタンパク質キニノーゲンを切断して、高分子量キニノーゲン(HMWK、フィッツジェラルド因子としても知られる)とブラジキニンを生成します。
Tissue-derived kallikrein has different properties than plasma kallikrein.
組織由来カリクレインは、血漿カリクレインとは異なる特性を持っています。
It is released physiologically at lower concentrations, mainly from exocrine glands, such as the salivary glands, kidney, and pancreas.
生理的には、唾液腺、腎臓、膵臓などの外分泌腺から低濃度で放出されます。
In addition, it can be synthesised by the activity of polycarboxypeptidase, also known as angiotensinase C, which is constitutively expressed on the surface of endothelial cells.
さらに、内皮細胞の表面に恒常的に発現するポリカルボキシペプチダーゼ(別名アンジオテンシナーゼ C)の活性によっても合成されます。
When activated (e.g., by pathogens), this enzyme converts plasma pre-kallikrein to kallikrein.
この酵素は活性化されると(例えば病原体によって)、血漿プレカリクレインをカリクレインに変換します。
Kininogens can also be cleaved by serine proteases other than kallikreins, such as human neutrophil elastase, tryptase, cathepsins, and proteinase-3.
キニノーゲンは、カリクレイン以外のセリンプロテアーゼ、例えばヒト好中球エラスターゼ、トリプターゼ、カテプシン、プロテイナーゼ-3によっても切断されます。
These processes also increase the production of kinins, such as Lys-bradykinin from L- and H- kininogens, which are rapidly bio-transformed to bradykinin via plasma aminopeptidases.
これらのプロセスは、L-キニノーゲンおよびH-キニノーゲンからのLys-ブラジキニンなどのキニンの産生も増加させ、これらは血漿アミノペプチダーゼを介してブラジキニンに急速に生体変換されます。
One of the main functions of bradykinin is to participate in inflammation, pain, and innate immunity.
ブラジキニンの主な機能の1つは、炎症、痛み、および自然免疫に関与することです。
Kallikreins are involved in important physiological functions, such as activation of the intrinsic pathway of blood coagulation, its regulation, and fibrinolysis.
カリクレインは、血液凝固の内因性経路の活性化、その調節、線溶などの重要な生理機能に関与しています。
In addition, bradykinin, HMWK, and FXII are involved in the contact system of innate immunity, which in turn participates in activation of the complement pathway (Figure 3).
さらに、ブラジキニン、HMWK、およびFXIIは、自然免疫の接触システムに関与しており、これが補体経路の活性化に関与しています(図3)。
The KKS is considered an extension of the RAAS.
KKS は RAAS の延長であると考えられています。
ACE has a greater affinity for bradykinin than for angiotensin II.
ACEはアンジオテンシンIIよりもブラジキニンに対して親和性が高い。
Thus, after kinin production, ACE2 preferentially degrades des-Arg9-bradykinin, whereas ACE preferentially metabolises bradykinin.
したがって、キニン生成後、ACE2 は des-Arg9-ブラジキニンを優先的に分解し、ACE はブラジキニンを優先的に代謝します。
Considering the rapid activity of ACE, the half-life of bradykinin and Lys-bradykinin is only 27 s, so these proteins act locally.
ACE の急速な活性を考慮すると、ブラジキニンと Lys-ブラジキニンの半減期はわずか 27 秒であるため、これらのタンパク質は局所的に作用します。
In addition, ACE bio-transforms 11% of bradykinin to des-Arg9-bradykinin, which increases its half-life ten-fold compared with bradykinin.
さらに、ACE はブラジキニンの 11% を des-Arg9-ブラジキニンに生体変換し、ブラジキニンと比較して半減期が 10 倍長くなります。
The different ACE isoforms, which have distinct activity, allow bradykinin to regulate angiotensin II activity and promote vasodilation, natriuresis, and hypotension through the bradykinin receptor B2.
異なる活性を持つ異なるACEアイソフォームにより、ブラジキニンはアンジオテンシンIIの活性を調節し、ブラジキニン受容体B2を介して血管拡張、ナトリウム利尿、低血圧を促進する。
These ACE isoforms exert peptidylpeptidase and kinase II activities and can metabolise kinins and kallikreins (Figure 3).
これらのACEアイソフォームはペプチジルペプチダーゼおよびキナーゼII活性を発揮し、キニンおよびカリクレインを代謝することができる(図3)。
Furthermore, angiotensin II increases B2 receptor expression, and angiotensin II receptor type 2 stimulates the expression of angiotensinase C in endothelial cells and, thus, the production of bradykinin.
さらに、アンジオテンシンIIはB2受容体の発現を増加させ、アンジオテンシンII受容体2型は内皮細胞におけるアンジオテンシナーゼCの発現を刺激し、ブラジキニンの産生を刺激する。
Regulation of the expression and activity of ACE and its isoforms is crucial.
ACEとそのアイソフォームの発現と活性の調節は極めて重要である。
When ACE2 is downregulated, there will be a preferential increase in des-Arg9-bradykinin levels.
ACE2がダウンレギュレーションされると、des-Arg9-ブラジキニンレベルが優先的に増加する。
As mentioned previously, ACE2 is downregulated in patients with COVID-19.
前述のように、COVID-19患者ではACE2の発現が低下している。
An increase in the ACE/ACE2 expression ratio has been associated with organ damage in these patients.
ACE/ACE2発現比の増加は、これらの患者の臓器障害と関連している。
Moreover, Roche and Roche suggested that plasma kinin concentrations should be monitored to help predict the severity of pulmonary problems.
さらに、ロシュとロシュは、肺疾患の重症度を予測するために血漿キニン濃度をモニタリングすべきであると示唆した。
In addition, kinins have been implicated in pain and inflammation that is potentiated by plasmin.
さらに、キニンはプラスミンによって増強される痛みや炎症に関与している。
Des-Arg9-bradykinin activates the B1 receptor with great affinity, leading to neutrophil recruitment, increased vascular permeability, and leucocyte extravasation into the lung.
Des-Arg9-ブラジキニンはB1受容体を高い親和性で活性化し、好中球の動員、血管透過性の増加、白血球の肺への血管外漏出を引き起こす。
Activation of this receptor leads to the release of chemokines, such as C-X-C motif chemokine 5 (CXCL5), from pulmonary epithelial cells, which in turn activates chemokine receptor type 2 (CXCR2) of neutrophils to facilitate their recruitment and infiltration into the lung and increasing inflammation.
この受容体が活性化されると、肺上皮細胞からC-X-Cモチーフケモカイン5(CXCL5)などのケモカインが放出され、好中球のケモカイン受容体2型(CXCR2)が活性化されて好中球の肺への動員と浸潤が促進され、炎症が増加する。
In these inflammatory conditions, the B1 receptor is upregulated almost 3000-fold and the B2 receptor is upregulated 200-fold compared with the normal state, resulting in respiratory distress syndrome and multiorgan failure.
これらの炎症状態では、B1受容体は正常状態と比較して約3000倍、B2受容体は200倍にアップレギュレーションされ、呼吸窮迫症候群および多臓器不全を引き起こします。
B1 and B2 receptor activity is highly sensitive to the action of cytokines and growth factors (e.g., IL-1β, IL-2, TNF-α, IFN-γ, and epidermal growth factor (EGF)) and toxins from microorganisms.
B1およびB2受容体の活性は、サイトカインや成長因子(IL-1β、IL-2、TNF-α、IFN-γ、上皮成長因子(EGF)など)および微生物由来の毒素の作用に非常に敏感です。
These effects, observed in patients with COVID-19, have also been detected in murine models with ACE2 downregulation.
COVID-19患者で観察されたこれらの効果は、ACE2ダウンレギュレーションを伴うマウスモデルでも検出されています。
Furthermore, in these models, ACE2 downregulation exacerbates inflammation and pulmonary oedema via increased B1 receptor activation by elevated Des-Arg9-bradykinin levels.
さらに、これらのモデルでは、ACE2ダウンレギュレーションは、Des-Arg9-ブラジキニンレベルの上昇によるB1受容体の活性化の増加を介して炎症と肺水腫を悪化させます。
The B2 receptor has also been linked to the production of fever, cough, bronchoconstriction, and increased airway resistance.
B2受容体は、発熱、咳、気管支収縮、気道抵抗の増加にも関連している。
It is also important to note that while plasma kininogen and kallikrein concentrations are very low in healthy individuals, very high values are detected in patients with COVID-19.
また、血漿キニノーゲンおよびカリクレイン濃度は健康な人では極めて低いが、COVID-19患者では非常に高い値が検出されることにも留意する必要がある。
Bradykinin also increases the activity of chymase, an endopeptidase released by mast cells to increase angiotensin II release and thus enhance the inflammatory process.
ブラジキニンはまた、肥満細胞から放出されるエンドペプチダーゼであるキマーゼの活性を高め、アンジオテンシンIIの放出を増加させ、炎症プロセスを強化します。
Bradykinin increases renin synthesis and release through the B2 receptor and induction of prostaglandin E2 release.
ブラジキニンは、B2受容体を介したレニンの合成と放出、およびプロスタグランジンE2の放出の誘導を増加させる。
In addition, tissue kallikreins can transform angiotensin I to angiotensin II.
さらに、組織カリクレインはアンジオテンシンIをアンジオテンシンIIに変換することができる。
In summary, bradykinin is substantially involved in the cytokine storm that occurs in patients with COVID-19 and in the various symptoms that occur during the course of this disease.
要約すると、ブラジキニンは、COVID-19 患者に発生するサイトカイン ストームと、この病気の経過中に発生するさまざまな症状に大きく関与しています。
Key points:
Bradykinin, HMWK, and FXII of coagulation form the backbone of the contact system of innate immunity and participate in complement activation.
ブラジキニン、HMWK、凝固因子 FXII は、自然免疫の接触システムのバックボーンを形成し、補体活性化に関与します。
The KKS is an extension of the RAAS. ACE has peptidylpeptidase and chymase II activity, so it not only metabolises angiotensin II but also kinases and kallikreins.
KKS は RAAS の延長です。ACE はペプチジルペプチダーゼとキマーゼ II 活性を持つため、アンジオテンシン II だけでなくキナーゼとカリクレインも代謝します。
The imbalance of the ACE/ACE2 ratio is associated with an increase in des-Arg9-bradykinin, which is related to neutrophilia, inflammation, and increased lung tissue damage.
ACE/ACE2 比の不均衡は、好中球増加、炎症、肺組織損傷の増加に関連する des-Arg9-ブラジキニンの増加と関連しています。
Des-Arg9-bradykinin is related to many of the symptomatic processes of COVID-19 (fever, cough, or bronchoconstriction).
des-Arg9-ブラジキニンは、COVID-19 の症状の多く (発熱、咳、気管支収縮) に関連しています。
By increasing chymase activity, bradykinin causes higher levels of angiotensin.
キマーゼ活性を高めることで、ブラジキニンはアンジオテンシンのレベルを高めます。
4.2. Involvement of the RAAS and the KKS in Thromboembolism
血栓塞栓症におけるRAASとKKSの関与
One of the most frequent causes of mortality after SARS-CoV-2 infection is the formation of micro-embolism and macro-embolism in the pulmonary and extrapulmonary vasculature.
SARS-CoV-2感染後の死亡原因として最も多いのは、肺血管および肺外血管における微小塞栓症および大塞栓症の形成である。
The causes of these events are related to vascular endothelial and epithelial cellular dysfunction that occurs after ACE2 internalisation.
これらの事象の原因は、ACE2 の内在化後に起こる血管内皮細胞および上皮細胞の機能不全に関連しています。
The phenomenon is related to three fundamental processes that occur after infection: (a) immune activation and production of proinflammatory cytokines by endothelial cells, (b) dysregulation of the RAAS, and (c) dysregulation of the KKS.
この現象は、感染後に起こる 3 つの基本的なプロセスに関連しています。(a) 内皮細胞による免疫活性化および炎症性サイトカインの産生、(b) RAAS の調節不全、(c) KKS の調節不全です。
In this section, we mainly discuss how RAAS and KKS dysregulation affect the thromboembolic processes.
このセクションでは、主に RAAS および KKS の調節不全が血栓塞栓プロセスにどのように影響するかについて説明します。
4.2.1. Immune Activation in Thromboembolism
血栓塞栓症における免疫活性化
Patients with COVID-19 present an exacerbated inflammatory response, known as the cytokine storm.
COVID-19 患者は、サイトカイン ストームとして知られる炎症反応の悪化を呈する。
Epithelial cells (especially pulmonary and vascular cells) play an important role in this phenomenon, leading to tissue damage and immuno-thrombosis.
上皮細胞(特に肺細胞と血管細胞)はこの現象に重要な役割を果たし、組織損傷と免疫血栓症を引き起こします。
This syndrome, which has a high incidence in patients with COVID-19 (10–20%), is characterised by high morbidity (e.g., micro-embolism, macro-embolism, and/or multiorgan failure) and lethality.
この症候群はCOVID-19患者で発生率が高く(10~20%)、高い罹患率(例:微小塞栓症、大塞栓症、および/または多臓器不全)と致死率が特徴です。
This situation is caused by expression of the viral proteins open reading frame 3b (ORF3b), ORF6 and ORF8 in infected cells.
この状況は、感染細胞におけるウイルスタンパク質のオープンリーディングフレーム3b(ORF3b)、ORF6、およびORF8の発現によって引き起こされます。
Together with the nucleocapsid (N) protein, these proteins are involved in facilitating rapid viral replication in epithelial cells (e.g., the vascular endothelium) and delaying or suppressing the response to type I and II IFNs, a process that involves NF-κB.
これらのタンパク質は、ヌクレオカプシド(N)タンパク質とともに、上皮細胞(血管内皮など)での急速なウイルス複製を促進し、NF-κBが関与するプロセスであるI型およびII型IFNへの応答を遅延または抑制することに関与しています。
Furthermore, ACE2 dysregulation occurs as a consequence of viral entry and the important roles that this enzyme has in immunity—including control of immune competence of myeloid cells and the clearance of peptides, such as AcSDKP, involved in activation of immunity, leading to dysregulated activity of CD8+ T lymphocytes, natural killer (NK) cells, and antigen-presenting cells.
さらに、ACE2 の調節不全は、ウイルスの侵入と、この酵素が免疫において果たす重要な役割(骨髄細胞の免疫能の制御、免疫活性化に関与する AcSDKP などのペプチドの除去など)の結果として発生し、CD8+ T リンパ球、ナチュラル キラー (NK) 細胞、抗原提示細胞の活動の調節不全につながります。
This causes a miscommunication between innate and adaptive immunity, inducing amplification of the cytokine-mediated inflammatory response, which is prolonged over time.
これにより、自然免疫と獲得免疫の間で誤ったコミュニケーションが生じ、サイトカインを介した炎症反応の増幅が誘発され、時間の経過とともに長引くことになります。
SARS-CoV-2 infection of endothelial cells causes dysfunction either directly, via viral activation of signalling pathways (e.g., the S protein binds to ACE2 to cause calcium-dependent toxic effects in endothelial cells), or indirectly by altering the endothelium-associated immune and inflammatory response.
内皮細胞のSARS-CoV-2感染は、シグナル伝達経路のウイルス活性化を介して直接的に機能不全を引き起こす(例えば、Sタンパク質がACE2に結合して内皮細胞にカルシウム依存性の毒性効果を引き起こす)、または内皮関連の免疫および炎症反応を変化させることによって間接的に機能不全を引き起こす。
This inappropriate response is also aggravated by infection of vascular basement membrane pericytes, which have high ACE2 expression.
この不適切な反応は、ACE2の発現が高い血管基底膜周皮細胞の感染によっても悪化する。
This leads to an imbalance in the endothelium–pericyte relationship, which has consequences in the signalling and the release of proinflammatory and profibrotic factors, such as angiopoietin I or platelet-derived growth factor.
これにより、内皮細胞と周皮細胞の関係に不均衡が生じ、シグナル伝達や、アンジオポエチンIや血小板由来成長因子などの炎症促進因子および線維化促進因子の放出に影響を及ぼします。
The combination of the correct production of these mediators and the degree of expression of their receptors, as well as pericyte death, triggers hypercoagulation, vascular permeability, oxidative stress, and the passage of toxins into adjacent tissues.
これらのメディエーターの正しい産生とその受容体の発現の程度、および周皮細胞の死が組み合わさって、過凝固、血管透過性、酸化ストレス、および隣接組織への毒素の通過を引き起こします。
This phenomenon has also been observed with SARS-CoV-2, as demonstrated in autopsies of patients with COVID-19.
この現象は、COVID-19患者の剖検で実証されているように、SARS-CoV-2でも観察されています。
SARS-CoV-2 also activates complement pathways, which are closely and reciprocally related to haemostasis.
SARS-CoV-2 は、止血と密接かつ相互に関連する補体経路も活性化します。
This activation may be due in part to endothelial damage and to the action of several SARS-CoV-2 structural proteins.
この活性化は、内皮損傷といくつかの SARS-CoV-2 構造タンパク質の作用に一部起因している可能性があります。
The N protein enhances the activation of the complement lectin pathway, and the S protein activates the alternative complement pathway by binding to heparan sulphate on cell surfaces and to C4a.
N タンパク質は補体レクチン経路の活性化を促進し、S タンパク質は細胞表面のヘパラン硫酸と C4a に結合することで代替補体経路を活性化します。
Both complement pathways converge with activation of C3a and C5a, anaphylatoxins that increase immune cell recruitment and ROS production, perpetuate endothelial damage, and cause thrombosis.
どちらの補体経路も、アナフィラトキシンであるC3aとC5aの活性化に収束し、免疫細胞の動員とROS産生を増加させ、内皮損傷を永続させ、血栓症を引き起こします。
C3a and C5a can stimulate the release of IL-6 and TNF from macrophages and other cells expressing the C3a/C5a receptors.
C3aとC5aは、マクロファージやC3a/C5a受容体を発現する他の細胞からのIL-6とTNFの放出を刺激することができる。
SARS-CoV-2 results in overexpression of these receptors, making these pathways more susceptible to activation.
SARS-CoV-2はこれらの受容体の過剰発現を引き起こし、これらの経路が活性化されやすくなります。
In addition, C5a can lead to the release of tissue factor and plasminogen activator inhibitor peptide type I (PAI-1) from endothelial cells.
さらに、C5aは内皮細胞からの組織因子およびプラスミノーゲン活性化因子阻害ペプチドI型(PAI-1)の放出を引き起こす可能性がある。
Key points:
SARS-CoV-2 causes an alteration in the endothelium–pericyte relationship, which leads to the release of inflammatory factors and cell death.
SARS-CoV-2 は内皮細胞と周皮細胞の関係に変化を引き起こし、炎症因子の放出と細胞死につながります。
SARS-CoV-2, through structural proteins, activates the contact system and the complement pathway. This causes the endothelium to release factors that participate in thrombo-inflammation, such as tissue factor or PAI-1.
SARS-CoV-2 は構造タンパク質を介して接触システムと補体経路を活性化します。これにより、内皮細胞は組織因子や PAI-1 など、血栓性炎症に関与する因子を放出します。
4.2.2. The RAAS in Thromboembolism
血栓塞栓症におけるRAAS
As mentioned previously, ACE downregulation is linked to the overexpression of ADAM17 that releases ACE2 anchored to the plasma membrane, increasing its soluble forms.
前述のように、ACE のダウンレギュレーションは、ADAM17 の過剰発現と関連しており、ADAM17 は細胞膜に固定された ACE2 を放出し、その可溶性形態を増加させます。
Since ACE2 regulates inflammation, its deregulation increases inflammation.
ACE2 は炎症を調節するため、その調節不全は炎症を増加させます。
One of the main culprits is ADAM17, which causes the release of proinflammatory factors such as TNF-α, TNF receptor 1 and 2, and IL-6R.
主な原因の 1 つは ADAM17 で、TNF-α、TNF 受容体 1 および 2、IL-6R などの炎症誘発因子の放出を引き起こします。
The release of soluble cytokine receptors and the formation of soluble complexes exacerbates the response through the JAK/STAT3 pathway.
可溶性サイトカイン受容体の放出と可溶性複合体の形成は、JAK/STAT3経路を介して反応を悪化させる。
This inflammatory state contributes to thromboembolic processes and increases angiotensin II levels, which also contribute to the inflammatory response and coagulation.
この炎症状態は血栓塞栓プロセスに寄与し、アンジオテンシン II レベルを上昇させます。アンジオテンシン II レベルは炎症反応と凝固にも寄与します。
Angiotensin II is a prothrombotic substance, as it increases the production of PAI-1 in endothelial cells.
アンジオテンシン II は、内皮細胞での PAI-1 の産生を増加させるため、血栓形成促進物質です。
Thus, an increase in angiotensin II may contribute to the local microthrombus formation in alveolar capillaries that occurs in patients with COVID-19, as fibrin is not degraded by tissue plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA).
したがって、フィブリンは組織プラスミノーゲン活性化因子(tPA)とウロキナーゼ型プラスミノーゲン活性化因子(uPA)によって分解されないため、アンジオテンシンIIの増加はCOVID-19患者に発生する肺胞毛細血管における局所的微小血栓形成に寄与している可能性がある。
Finally, it is important to note once again that there is cooperation between the RAAS and the KKS.
最後に、RAAS と KKS の間には協力関係があることを改めて指摘しておくことが重要です。
Due to RAAS deregulation, there is greater production of kinins, which also contribute to perturb haemostasis in patients with COVID-19.
RAAS の調節解除によりキニンの生成が増加し、これも COVID-19 患者の止血を乱す一因となります。
4.2.3. The KKS in Thromboembolism
血栓塞栓症におけるKKS
The main contact system components are FXII, HMWK, and pre-kallikrein, which circulate in the blood in the form of zymogens.
接触系の主な成分は、酵素原の形で血液中を循環するFXII、HMWK、プレカリクレインである。
They are activated after binding to antigenic molecules of pathogens with high structural variability, such as nucleic acids of microorganisms, NETs, and ferritin.
これらは、微生物の核酸、NET、フェリチンなど、構造的に変動性の高い病原体の抗原分子に結合した後に活性化される。
Numerous SARS-CoV-2 antigens (e.g., structural proteins, such as S1, N, M, and E) have a high capacity to bind to complement proteins and the contact system, leading to their activation.
多数のSARS-CoV-2抗原(例えば、S1、N、M、Eなどの構造タンパク質)は、補体タンパク質や接触系に結合する能力が高く、それらの活性化につながる。
Furthermore, remnants of these antigens after infection may continue to activate these innate immunity pathways, which might contribute to the symptoms in patients with long COVID.
さらに、感染後に残ったこれらの抗原は、これらの自然免疫経路を活性化し続ける可能性があり、それが長期COVID患者の症状に寄与している可能性がある。
The binding of these antigens to proteins of the contact system (HMWK and pre-kallikrein) triggers, through FXII, activation of the intrinsic pathway of blood coagulation and the KKS, producing plasma kallikrein and bradykinin that provide feedback to FXII activation.
これらの抗原が接触システムのタンパク質(HMWKおよびプレカリクレイン)に結合すると、FXIIを介して血液凝固の内因性経路とKKSが活性化され、FXII活性化にフィードバックを提供する血漿カリクレインとブラジキニンが生成されます。
In addition, the contact system can be activated independently of FXII activity, including through angiotensinase C from endothelial cells or increased release of proteases (e.g., trypsin) from glandular tissue in response to infection.
さらに、接触系は、感染に反応して内皮細胞からのアンジオテンシナーゼCや腺組織からのプロテアーゼ(トリプシンなど)の放出増加などを通じて、FXII活性とは独立して活性化される可能性がある。
In COVID-19, the levels and activity of plasma kallikreins and kininogens are increased.
COVID-19では、血漿カリクレインとキニノーゲンのレベルと活性が増加します。
This increased kallikrein activity causes consumption of intrinsic coagulation pathway factors, an increase in the activated partial thromboplastin time, conversion of plasminogen to plasmin, and a state of hyperfibrinolysis.
このカリクレイン活性の増加は、内因性凝固経路因子の消費、活性化部分トロンボプラスチン時間の増加、プラスミノーゲンからプラスミンへの変換、および過剰線溶状態を引き起こす。
Plasmin increases the production of bradykinin, contributing to an inflammatory state.
プラスミンはブラジキニンの産生を増加させ、炎症状態に寄与する。
Bradykinin also contributes to this inflammatory and hyperfibrinolytic state through the release of tPA, implicating it in the coagulation imbalances that occur in patients with COVID-19 (hyperfibrinolysis or thromboembolism).
ブラジキニンはtPAの放出を通じてこの炎症性および過剰線溶状態に寄与し、COVID-19患者に生じる凝固不均衡(過剰線溶または血栓塞栓症)に関与している。
In addition, plasmin and FXII can be reciprocally activated.
さらに、プラスミンとFXIIは相互に活性化される可能性がある。
The pathological increase in FXII contributes not only to septic phenomena and blood dyscrasias, but also to fibroblast proliferation and pulmonary fibrosis, phenomena that occur in patients with COVID-19.
FXIIの病理学的増加は敗血症や血液疾患だけでなく、COVID-19患者に起こる線維芽細胞の増殖や肺線維症にも寄与する。
Key points:
Angiotensin II is a factor of thrombo-inflammation in the pulmonary blood capillaries, by increasing the production of PAI-1.
アンジオテンシン II は、PAI-1 の産生を増加させることによって、肺毛細血管の血栓性炎症の要因となります。
Binding of SARS-CoV-2 antigens to the contact system activates the intrinsic coagulation pathway and KKS.
SARS-CoV-2 抗原が接触系に結合すると、内因性凝固経路と KKS が活性化されます。
The increase in protease activity (e.g., kallikreins or PAR receptors) causes a state of hyperfibrinolysis through the release of tPA. This participates in septic phenomena, blood dyscrasias, fibroblast proliferation, and pulmonary fibrosis.
プロテアーゼ活性 (カリクレインや PAR 受容体など) の増加により、tPA の放出によって線溶亢進状態が引き起こされます。これは、敗血症、血液疾患、線維芽細胞の増殖、および肺線維症に関与します。
5. Conclusions
SARS-CoV-2 uses plasma membrane and soluble proteases in its infective mechanism.
SARS-CoV-2 は、感染メカニズムに細胞膜プロテアーゼと可溶性プロテアーゼを使用します。
The way it enters the host cell may vary depending on which one is used (e.g., trypsin or cathepsins).
宿主細胞への侵入方法は、使用されるもの (トリプシンまたはカテプシンなど) によって異なります。
In addition, proteases also participate in the replication and maturation of viral proteins, as well as the release of new virions.
さらに、プロテアーゼはウイルスタンパク質の複製と成熟、および新しいウイルス粒子の放出にも関与します。
Upon viral entry, proteases cleave the S protein to anchor to the host cell via ACE2.
ウイルスが侵入すると、プロテアーゼが S タンパク質を切断し、ACE2 を介して宿主細胞に固定します。
This event causes dysregulation of ACE2, increasing its soluble forms and decreasing the one anchored in the cell membrane, a process that is mediated by ADAM17.
このイベントにより ACE2 の調節異常が起こり、可溶性形態が増加し、細胞膜に固定された形態が減少します。このプロセスは ADAM17 によって媒介されます。
This has important consequences on inflammation.
これは炎症に重要な影響を及ぼします。
The main consequence of ACE2 dysregulation is the increase in the ACE/ACE2 ratio.
ACE2 の調節不全の主な結果は、ACE/ACE2 比の増加です。
Because RAAS is an extension of KKS, the release and activity of proteases (e.g., kallikreins) will be unbalanced against the anti-proteases that regulate them.
RAAS は KKS の延長であるため、プロテアーゼ (カリクレインなど) の放出と活性は、それらを制御する抗プロテアーゼに対して不均衡になります。
These are going to activate each other in a chain.
これらは連鎖的に互いに活性化します。
This imbalance contributes to the infectious and proinflammatory mechanism, as well as to the imbalance of respiratory function.
この不均衡は、感染および炎症誘発メカニズム、ならびに呼吸機能の不均衡に寄与します。
Furthermore, KKS is part of the contact system of innate immunity together with the complement system and activates the intrinsic coagulation pathway.
さらに、KKS は補体系とともに自然免疫の接触システムの一部であり、内因性凝固経路を活性化します。
Angiotensin II and des-Arg9-bradykinin are going to be the two main final mediators of this entire chain of events.
アンジオテンシン II と des-Arg9-ブラジキニンは、この一連のイベント全体の 2 つの主な最終メディエーターになります。
Among the consequences of this will be the affectation of the endothelium–pericyte relationship, fibroblast proliferation, the death of the vascular endothelium, hyperinflammation, and the release of procoagulant factors, which in the most severe cases cause hyperfibrinolysis, thrombosis, and sepsis.
その結果として、内皮細胞と周皮細胞の関係への影響、線維芽細胞の増殖、血管内皮の死、過炎症、および凝血促進因子の放出が起こり、最も重篤なケースでは、過剰線維素溶解、血栓症、および敗血症を引き起こします。
These protease-mediated mechanisms are critical to understanding COVID-19 disease.
これらのプロテアーゼを介したメカニズムは、COVID-19 疾患を理解する上で重要です。
These mechanisms have not been explained in depth previously.
これらのメカニズムはこれまで詳細に説明されていませんでした。
Aprotinin is a broad-spectrum inhibitor of the most important proteases involved in SARS-CoV-2 infection.
アプロチニンは、SARS-CoV-2 感染に関与する最も重要なプロテアーゼの広域スペクトル阻害剤です。
We describe its pharmacodynamics, pharmacokinetics, toxicity, and potential for the treatment of various respiratory viruses in a second-part review, entitled “Aprotinin (II): Inhalational Administration for the Treatment of COVID-19 and Other Viral Conditions”.
第2部のレビュー「アプロチニン(II):COVID-19およびその他のウイルス性疾患の治療のための吸入投与」では、その薬力学、薬物動態、毒性、およびさまざまな呼吸器ウイルスの治療における可能性について説明します。
以下省略。
この記事が気に入ったらサポートをしてみませんか?