SARS-CoV-2 ヌクレオカプシドタンパク質の構造と組み立ての可塑性

Plasticity in structure and assembly of SARS-CoV-2 nucleocapsid protein
SARS-CoV-2 ヌクレオカプシドタンパク質の構造と組み立ての可塑性
元→Plasticity in structure and assembly of SARS-CoV-2 nucleocapsid protein | bioRxiv
Abstract
Worldwide SARS-CoV-2 sequencing efforts track emerging mutations in its spike protein, as well as characteristic mutations in other viral proteins.
世界中で SARS-CoV-2 の配列解析が行われ、スパイクタンパク質の新たな変異や、他のウイルスタンパク質の特徴的な変異が追跡されています。
Besides their epidemiological importance, the observed SARS-CoV-2 sequences present an ensemble of viable protein variants, and thereby a source of information on viral protein structure and function.
疫学的重要性に加えて、観察された SARS-CoV-2 配列は、生存可能なタンパク質変異体の集合体であり、ウイルスタンパク質の構造と機能に関する情報源となります。
Charting the mutational landscape of the nucleocapsid (N) protein that facilitates viral assembly, we observe variability exceeding that of the spike protein, with more than 86% of residues that can be substituted, on average by 3-4 different amino acids.
ウイルスの組み立てを促進するヌクレオカプシド (N) タンパク質の変異ランドスケープを図表化すると、スパイクタンパク質を超える変動性が見られ、残基の 86% 以上が平均して 3~4 個の異なるアミノ酸に置換できます。
However, mutations exhibit an uneven distribution that tracks known structural features but also reveals highly protected stretches of unknown function.
しかし、変異は不均一な分布を示し、既知の構造的特徴をたどる一方で、高度に保護された未知の機能領域も明らかにしています。
One of these conserved regions is in the central disordered linker proximal to the N-G215C mutation that has become dominant in the Delta variant, outcompeting G215 variants without further spike or N-protein substitutions.
これらの保存領域の 1 つは、Delta バリアントで優勢になった N-G215C 変異の近位にある中央の無秩序なリンカーにあり、スパイクや N タンパク質のさらなる置換なしに G215 バリアントに打ち勝ちました。
Structural models suggest that the G215C mutation stabilizes conserved transient helices in the disordered linker serving as protein-protein interaction interfaces.
構造モデルは、G215C 変異が、無秩序なリンカー内の保存された一時的なヘリックスを安定化し、タンパク質間相互作用インターフェイスとして機能することを示唆しています。
Comparing Delta variant N-protein to its ancestral version in biophysical experiments, we find a significantly more compact and less disordered structure.
生物物理学的実験でデルタ変異体 N タンパク質をその祖先バージョンと比較すると、構造がはるかにコンパクトで乱れが少ないことがわかります。
N-G215C exhibits substantially stronger self-association, shifting the unliganded protein from a dimeric to a tetrameric oligomeric state, which leads to enhanced co-assembly with nucleic acids.
N-G215C は、かなり強力な自己会合を示し、リガンドのないタンパク質を二量体から四量体のオリゴマー状態に移行させ、核酸との共集合を強化します。
This suggests that the sequence variability of N-protein is mirrored by high plasticity of N-protein biophysical properties, which we hypothesize can be exploited by SARS-CoV-2 to achieve greater efficiency of viral assembly, and thereby enhanced infectivity.
これは、N タンパク質の配列の多様性が N タンパク質の生物物理学的特性の高い可塑性に反映されていることを示唆しており、SARS-CoV-2 はこれを利用してウイルスの集合効率を高め、感染力を高めることができると仮定しています。
Introduction
Two years into the COVID19 pandemic, intense research into the structure and molecular mechanisms of SARS-CoV-2 virus has led to the rapid development and deployment of several types of vaccines, neutralizing monoclonal antibodies, and small molecules drugs such as ribonucleoside analogs and inhibitors of viral proteases and polymerase.
COVID19パンデミックから2年が経ち、SARS-CoV-2ウイルスの構造と分子メカニズムに関する熱心な研究により、数種類のワクチン、中和モノクローナル抗体、リボヌクレオシド類似体、ウイルスプロテアーゼおよびポリメラーゼの阻害剤などの低分子薬が急速に開発され、導入されました。
A persistent concern is viral escape through evolution of therapeutic and immunological targets.
治療および免疫学的標的の進化によるウイルスの逃避は、根強い懸念事項です。
Most attention in this regard is devoted to the viral spike protein that facilitates viral entry, though recent data additionally point to the importance of viral packaging by the nucleocapsid (N) protein modulating viral loads and thereby infectivity.
この点で最も注目されているのは、ウイルスの侵入を促進するウイルススパイクタンパク質ですが、最近のデータでは、ウイルス量と感染性を調節するヌクレオカプシド (N) タンパク質によるウイルスパッケージングの重要性も指摘されています。
In an unprecedented global effort, several millions of genomes have been sequenced so far and submitted to the Global Initiative on Sharing All Influenza Data (GISAID) to monitor SARS-CoV-2 variants (Fig. 1a).
前例のない世界的な取り組みにより、これまでに数百万のゲノムが配列決定され、SARS-CoV-2 変異体の監視のために、インフルエンザデータ共有に関する世界イニシアチブ (GISAID) に提出されました (図 1a)。
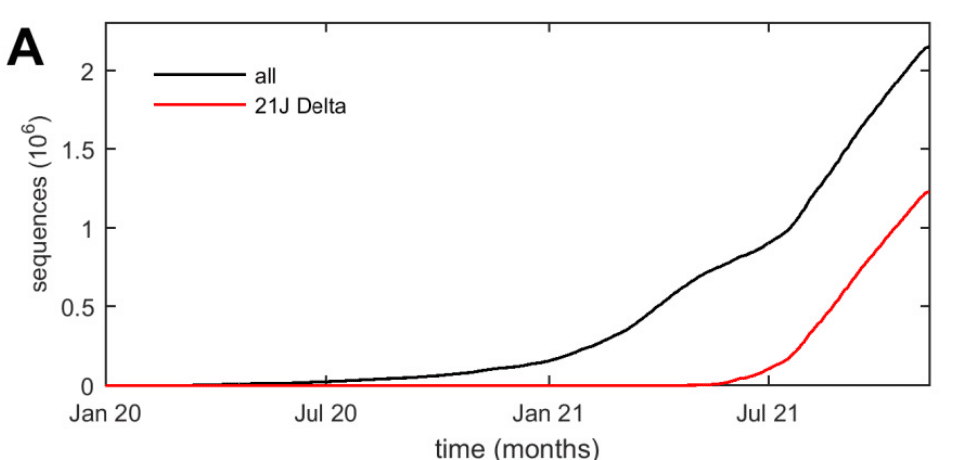
This has provided an invaluable data base for recognizing emerging variants of concern, phylogenetic analyses, and analyses of geographic spread.
これにより、懸念される新たな変異体の認識、系統解析、地理的拡散の解析のための貴重なデータベースが提供されました。
The vast majority of the observed mutations are short-lived and ostensibly inconsequential.
観察された変異の大部分は短命で、一見重要ではない。
However, such mutations play a key role in establishing the genetic diversity of RNA viruses, with profound impact on evolutionary dynamics.
しかし、このような変異はRNAウイルスの遺伝的多様性を確立する上で重要な役割を果たし、進化のダイナミクスに大きな影響を与える。
In accumulation they define a mutational landscape that is intimately related to stability constraints and structure-function relationships of viral proteins.
蓄積すると、ウイルスタンパク質の安定性制約と構造機能関係に密接に関連する変異ランドスケープが定義されます。
Thus, projecting the ensemble of mutations in reported sequences into the amino acid sequence space and neglecting their origin and relationships, they manifest an exhaustive mutational landscape of viable amino acid substitutions of SARS-CoV-2 proteins, all of which evidently have been successfully proliferating as part of the SARS-CoV-2 species found in patients.
したがって、報告された配列の変異の集合をアミノ酸配列空間に投影し、その起源と関係を無視すると、SARS-CoV-2タンパク質の生存可能なアミノ酸置換の網羅的な変異ランドスケープが明らかになります。 これらはすべて、患者に見られるSARS-CoV-2種の一部としてうまく増殖していることが明らかです。
Unfortunately, the fine-grained interpretation of such mutation data in the biophysical context of protein structure and function is a daunting task, and the spectrum of biophysical properties defined by the ensemble of mutant sequences is largely unclear, although it has been observed that RNA virus proteins generally have more loosely packed cores and intrinsically disordered regions that may provide adaptability.
残念ながら、タンパク質の構造と機能の生物物理学的文脈におけるこのような変異データのきめ細かな解釈は困難な作業であり、変異配列の集合によって定義される生物物理学的特性の範囲はほとんど不明であるが、RNAウイルスタンパク質は一般に、より緩く詰まったコアと、適応性をもたらす可能性のある本質的に無秩序な領域を持っていることが観察されている。
Focused analyses of RNA virus mutational landscapes in relation to protein structures have been carried out, for example, for the poliovirus polymerase, and the SARS-CoV-2 spike protein receptor binding domain.
ポリオウイルスポリメラーゼや SARS-CoV-2 スパイクタンパク質受容体結合ドメインなど、タンパク質構造に関連した RNA ウイルスの変異ランドスケープの集中的な分析が行われてきました。
In the present work we aim to exploit the mutational landscape of the SARS-CoV-2 N protein as a tool to elucidate molecular aspects of the viral assembly, which requires packaging the RNA through an as-of-yet incompletely understood co-assembly mechanism with N protein into well-defined ribonucleoprotein particles.
本研究では、SARS-CoV-2 Nタンパク質の変異ランドスケープを、ウイルスアセンブリの分子的側面を解明するためのツールとして活用することを目指しています。これには、Nタンパク質との共アセンブリメカニズムを通じてRNAをパッケージ化し、明確に定義されたリボ核タンパク質粒子にする必要がありますが、これはまだ完全には理解されていません。
Of particular interest with regard to viral assembly is the SARS-CoV-2 variant B.1.617.2 (Delta) that in 2021 has rapidly outperformed all previous variants (Fig. 1a), exhibiting reduced incubation time and significantly higher viral load in infected patients, in one study up to a 1000-fold higher compared to the original lineage.
ウイルスの組み立てに関して特に興味深いのは、2021年にこれまでのすべての変異株を急速に上回ったSARS-CoV-2変異株B.1.617.2(デルタ)です(図1a)。潜伏期間の短縮と感染患者のウイルス量の大幅な増加が見られ、ある研究では元の系統と比較して最大1000倍にまで増加しました。
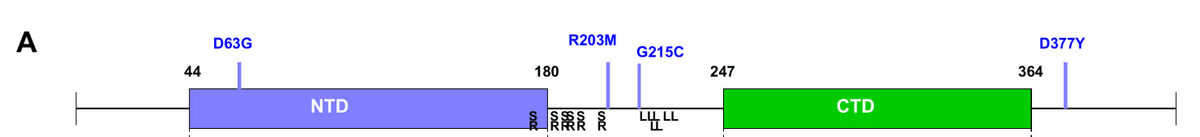
Delta variant mutations in N-protein include D63G, R203M, D377Y, and in different clades additionally R385K and G215C, respectively.
Nタンパク質のデルタ変異体変異には、それぞれD63G、R203M、D377Y、および異なる系統ではさらにR385KとG215Cが含まれます。
Among those, mutation R203M was shown to significantly increase replication, but is similar to analogous N-protein mutations in all other SARS-CoV-2 variants of concern including the current Omicron variant.
その中で、変異 R203M は複製を大幅に増加させることが示されましたが、現在のオミクロン変異体を含む懸念される他のすべての SARS-CoV-2 変異体の類似の N タンパク質変異と類似しています。
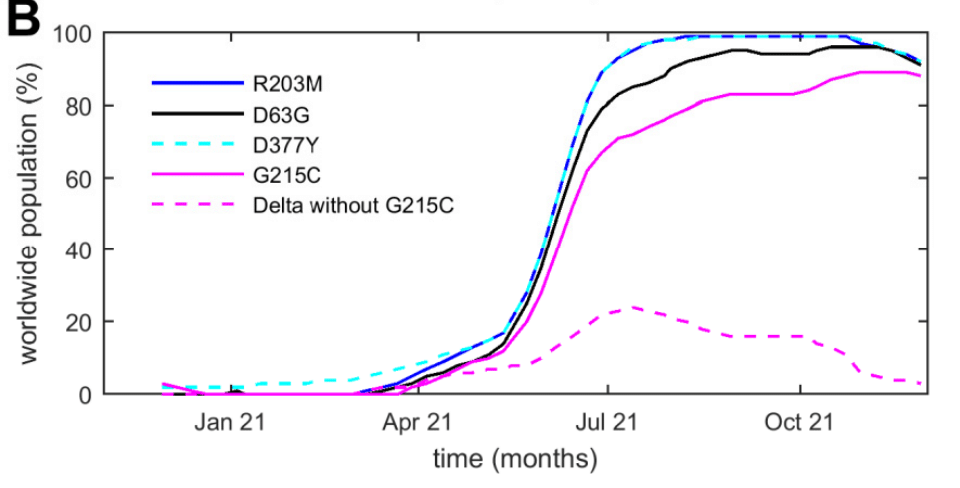
Conspicuously, the Delta variant containing the G215C mutation arose without accompanying changes in the spike protein (Nextstrain clade 21J) and has dramatically outperformed and virtually displaced other Delta variant clades, assuming worldwide dominance in 2021(Fig. 1b).
注目すべきことに、G215C 変異を含む Delta 変異体は、スパイクタンパク質の変化を伴わずに発生し (Nextstrain クレード 21J)、他の Delta 変異クレードを大幅に上回り、事実上それらを置き換え、2021 年には世界中で優位に立つと想定されています (図 1b)。
This warrants a detailed study of the impact of the G215C mutation on protein structure and function.
これは、G215C 変異がタンパク質の構造と機能に与える影響の詳細な研究を正当化します。
In the present work, we observe a highly conserved region that reveals a possible role of the G215C mutation in enhancing interactions critical for assembly.
本研究では、高度に保存された領域を観察し、アセンブリに不可欠な相互作用を強化する上での G215C 変異の可能性のある役割を明らかにしました。
To examine molecular mechanisms in detail, we combine biophysical characterization of protein size, shape, structure, and protein interactions with structural models from molecular dynamics simulations.
分子メカニズムを詳細に調べるために、タンパク質のサイズ、形状、構造、およびタンパク質相互作用の生物物理学的特性と、分子動力学シミュレーションからの構造モデルを組み合わせました。
In comparison with biophysical properties of the ancestral N-protein, the G215C mutant displays significant differences in secondary structure, self-association and co-assembly with nucleic acid, revealing a plasticity of protein biophysical properties that mirrors its remarkable sequence variability.
祖先 N タンパク質の生物物理学的特性と比較すると、G215C 変異体は二次構造、自己会合、および核酸との共アセンブリに大きな違いを示し、その顕著な配列可変性を反映するタンパク質の生物物理学的特性の可塑性を明らかにしています。
Results
The Mutational Landscape of SARS-CoV-2 N-protein Reflects Structural Features
SARS-CoV-2 Nタンパク質の変異地形は構造的特徴を反映
Among 2.49 million SARS-CoV-2 sequences uploaded to GISAID and subsequently preprocessed at Nextstrain.org as of November 29, 2021, we find 8.2 million instances of amino acid changes in the N-protein.
2021年11月29日現在、GISAIDにアップロードされ、その後Nextstrain.orgで前処理された249万件のSARS-CoV-2配列のうち、Nタンパク質のアミノ酸変化の例は820万件あります。
Outside the consensus mutations associated with different clades (see below), each sequence exhibits on average only 1.3 additional mutations in N-protein.
異なる系統群に関連するコンセンサス変異(下記参照)以外では、各配列は平均してNタンパク質の追加変異を1.3件のみ示しています。
From inspection of phylogenetic trees at Nextstrain.org, a majority of mutations arose multiple times independently, but usually persisted only briefly.
Nextstrain.orgの系統樹の検査から、変異の大部分は独立して複数回発生しましたが、通常は短期間しか持続しませんでした。
Despite their sparsity, when aggregated over 10⁶ sequences, the mutation data is highly redundant, describing 1,264 distinct mutations observed a median of 70 times, assembled in different combinations within 24,982 distinct N-protein sequences (Fig. 2).
それらの希薄さにもかかわらず、 10⁶ 配列にわたって集約すると、変異データは非常に冗長であり、24,982 の異なる N タンパク質配列内でさまざまな組み合わせで組み立てられた、平均 70 回観察された 1,264 の異なる変異を説明しています (図 2)。
Mutations occurred for 362 out of 419 residues of the N-protein, each on average allowing 3.5 different substitutions.
Nタンパク質の419残基のうち362残基に変異が発生し、それぞれ平均3.5の異なる置換が可能でした。
In range and depth of variability, N-protein exceeds all other structural SARS-CoV-2 proteins as well as ORF1ab (SI Appendix Table S1).
変異の範囲と深さにおいて、Nタンパク質は他のすべてのSARS-CoV-2構造タンパク質およびORF1abを上回っています(SI付録表S1)。
Notably, this plasticity of the amino acid sequence does not extend to most of the 37 positions strictly conserved across related betacoronaviruses, 30 of which exhibited no or only conservative substitutions in SARS-CoV-2 (Fig. 2).
注目すべきことに、アミノ酸配列のこの可塑性は、関連するベータコロナウイルス全体で厳密に保存されている37の位置のほとんどには及んでおらず、30の位置はSARS-CoV-2でまったく置換されていないか、保存的置換のみを示しています(図2)。
To assess to what extent the observed sequence space has exhausted the range of possible substitutions, Fig. 1c shows the scope of residues at which mutations have been observed as a function of total sequences.
観察された配列空間がどの程度まで可能な置換の範囲を使い果たしたかを評価するために、図 1c は、変異が観察された残基の範囲を全配列の関数として示しています。
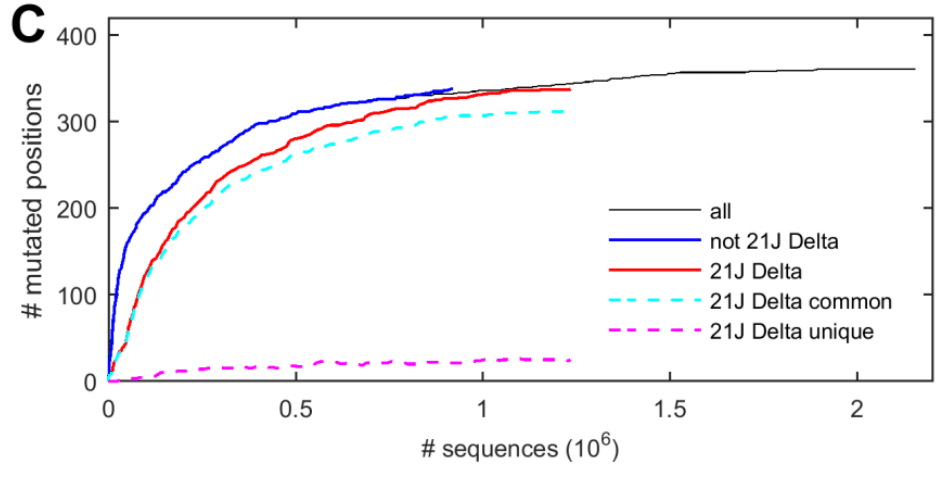
(Because we assume most sequence fluctuations to be stochastic independent events, the cumulative number of sequences is serving here as a scaled surrogate for time to compensate for vastly different sequence deposition rates with time.)
(ほとんどの配列変動は確率的独立イベントであると想定しているため、ここでは配列の累積数が時間の尺度化された代理として機能し、時間とともに大きく異なる配列沈着率を補正します。)
The approach of an asymptotic limit can be discerned, with a second phase of changes in emerging Delta variant sequences; these, too, approach a limit.
漸近的限界への接近が認められ、デルタ変異体配列の出現における第 2 段階の変化が見られます。これらも限界に近づいています。
This suggests that the observed mutation landscape approximates an equilibrium, while hinting at some changes associated with the Delta variant.
これは、観察された変異ランドスケープが平衡に近づいていることを示唆していますが、Delta 変異体に関連するいくつかの変化を示唆しています。
To further analyze the time course of observed mutations, Fig. 3e presents the temporal evolution of the mutation frequency observed at each position.
観察された変異の経時的変化をさらに分析するために、図 3e は各位置で観察された変異頻度の時間的変化を示しています。
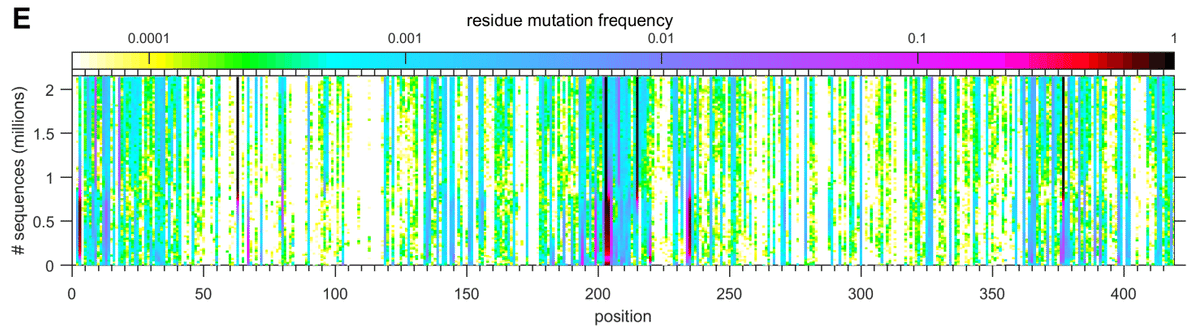
Insofar as the propagation of mutations in each N-protein position are driven by adventitious spreading events, or are bystander of improvements in fitness to other viral proteins, variation along the ordinate (time/sequence axis) in Fig. 3e is produced.
各 N タンパク質位置での変異の伝播が偶発的な拡散イベントによって引き起こされるか、または他のウイルスタンパク質への適応性の向上の傍観者である限り、図 3e の縦軸 (時間/シーケンス軸) に沿った変化が生成されます。
By contrast, constant mutation frequencies in individual residues (constant color along vertical line in Fig. 3e) must reflect intrinsic molecular properties.
対照的に、個々の残基における一定の変異頻度(図 3e の縦線に沿った一定の色)は、固有の分子特性を反映しているに違いありません。
Remarkably, for the overwhelming majority of positions across the N-protein, the observed mutation frequency is largely constant with minor stochastic modulations; this becomes even clearer when subdividing sequences between Delta 21J clades and others pre-dating 21J (SI Appendix Fig. S1).
驚くべきことに、N タンパク質全体の圧倒的多数の位置では、観測された変異頻度は、わずかな確率的変動を伴いほぼ一定です;これは、デルタ 21J 系統と 21J より前の系統の間で配列を細分化するとさらに明確になります (SI 付録図 S1)。
However, from the observed time-course of ancestral SARS-CoV-2 and Delta 21J mutations, it did require on the order of 10⁵ sequences to approximate the mutational landscape (SI Appendix Fig. S2).
ただし、祖先 SARS-CoV-2 とデルタ 21J の変異の観測された時間経過から、変異ランドスケープを近似するには 10⁵ のオーダーの配列が必要でした (SI 付録図 S2)。
We can better examine the significance of the mutations in more detail in the context of the N-protein structural organization and its assembly function.
N タンパク質の構造組織とそのアセンブリ機能の文脈で、変異の重要性をより詳細に調べることができます。
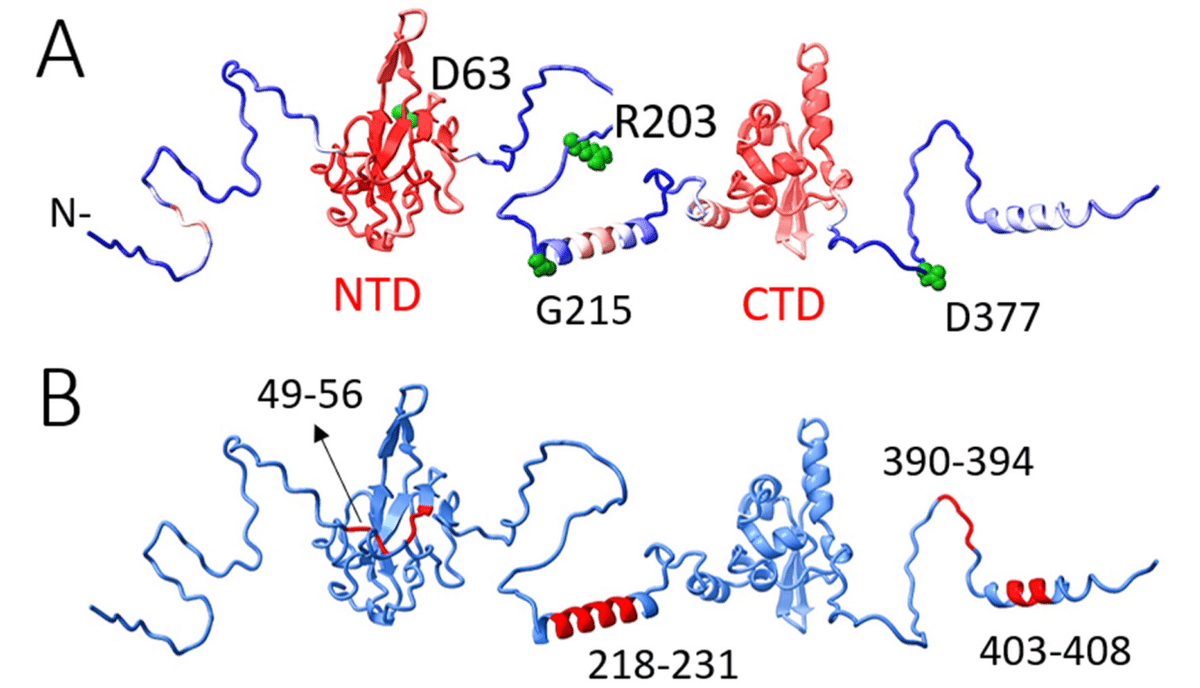
Briefly, N-protein is dimeric, with each chain comprised of a C-terminal dimerization domain (CTD) and an N-terminal nucleic acid (NA) binding domain (NTD), both flanked and linked by long intrinsically disordered arms (Fig. 3a).
簡単に言うと、N タンパク質は二量体であり、各鎖は C 末端二量体化ドメイン (CTD) と N 末端核酸 (NA) 結合ドメイン (NTD) で構成され、両方とも長い本質的に無秩序なアームによって挟まれて連結されています (図 3a)。
Both domains and linker contribute to promiscuous NA binding.
ドメインとリンカーの両方が、無差別な NA 結合に寄与しています。
NA binding induces a more ordered conformation that allows dimer-dimer interactions, which, in turn, and in concert with scaffolding on NA, lead to multimeric co-assemblies.
NA 結合は、二量体間相互作用を可能にするより秩序だった構造を誘導し、それが今度は NA 上の足場と相まって、多量体共集合につながります。
NA binding also promotes liquid-liquid phase separation (LLPS), and the highly concentrated co-condensates of N-protein and NA allow the formation of ribonucleoprotein particles.
NA 結合は液液相分離 (LLPS) も促進し、N タンパク質と NA の高濃度共凝縮物によりリボ核タンパク質粒子の形成が可能になります。
N-protein also interacts with the viral M-protein, which appears to play a role in promotion of N-protein condensates, in anchoring ribonucleoprotein particles to the viral membrane, and in the recognition of viral RNA.
N タンパク質はウイルスの M タンパク質とも相互作用し、N タンパク質凝縮物の促進、リボ核タンパク質粒子のウイルス膜への固定、およびウイルス RNA の認識に役割を果たしているようです。
Overall, the number of distinct mutations tracks well with the folded domains, which have a more restricted range of viable mutations (Fig. 3c).
全体的に、異なる変異の数は、実行可能な変異の範囲がより限定されている折り畳みドメインとよく一致しています (図 3c)。
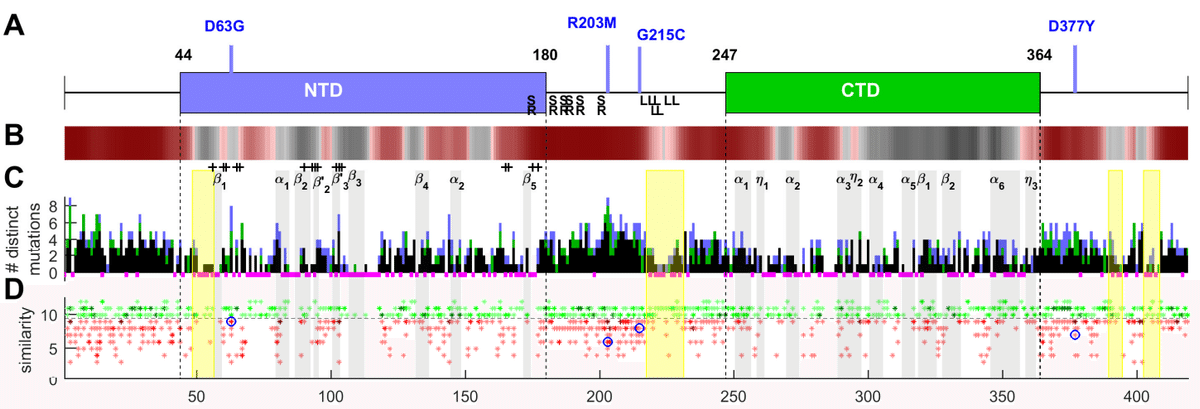
Residues close to the secondary structure elements previously revealed by NMR spectroscopy and x-ray crystallography are mostly either completely protected from mutations or highly conserved, as are the majority of NTD residues likely in contact with nucleic acids, as inferred from NMR chemical shifts.
NMR 分光法と X 線結晶構造解析によって以前に明らかにされた二次構造要素に近い残基は、ほとんどが変異から完全に保護されているか、高度に保存されています。これは、NMR 化学シフトから推測されるように、核酸と接触している可能性が高い NTD 残基の大部分も同様です。
Conversely, regions in the linker that score high for disorder and LLPS propensity (Fig. 3b) exhibit the largest clusters of mutations.
逆に、リンカー内の無秩序性と LLPS 傾向のスコアが高い領域 (図 3b) は、変異の最大のクラスターを示しています。
When examining the nature of the replacements through a scoring for physicochemical properties, such as polarity, hydrophobicity, and secondary structure propensity, we can discern a majority of conservative substitutions in the folded domains, and greatest variety of residues with altered physicochemical properties in the disordered regions (Fig. 3d).
極性、疎水性、二次構造傾向などの物理化学的特性のスコアリングを通じて置換の性質を調べると、折り畳まれたドメインで大部分の保守的な置換が見られ、無秩序な領域では物理化学的特性が変化した残基の種類が最も多いことがわかります (図 3d)。
New features appear in this mutational landscape in the disordered linkers and arms.
この変異ランドスケープでは、不規則なリンカーとアームに新たな特徴が見られます。
The linker is critical to RNA-mediated phase separation and was early identified as a region of high sequence variability.
リンカーは RNA を介した相分離に不可欠であり、配列の変動性が高い領域として早期に特定されました。
The large coverage of the mutational landscape now available allows identification of constrained islands within the disordered regions.
現在利用可能な変異ランドスケープの広範なカバレッジにより、不規則領域内の制約されたアイランドを特定できます。
These include positions 49–56 proximal to the first sheet in the NTD, the highly conserved leucine-rich sequence 218–231, as well as two stretches in the C-terminal arm (C-arm) at 390–394 and 403–408 (yellow highlights in Fig. 3c, d).
これらには、NTD の最初のシートに近い位置 49~56、高度に保存されたロイシンに富む配列 218~231、および C 末端アーム (C アーム) の 390~394 と 403~408 の 2 つの領域 (図 3c、d の黄色のハイライト) が含まれます。
Interestingly, these regions also approximately correspond to distinct stretches of amino acids with low disorder score.
興味深いことに、これらの領域は、不規則スコアが低いアミノ酸の明確な領域にもほぼ対応しています。
Based on their apparent conservation, these regions likely endow N-protein with critical functions.
明らかに保存されていることから、これらの領域は N タンパク質に重要な機能を与えていると考えられます。
The disordered C-arm of N-protein is thought to play a critical role in the interaction with viral M-protein and the packaging signal, based on studies with corresponding regions of other coronaviruses.
他のコロナウイルスの対応する領域の研究に基づくと、N タンパク質の不規則な C アームは、ウイルスの M タンパク質およびパッケージング シグナルとの相互作用において重要な役割を果たすと考えられています。
The observed conserved stretches in the mutational landscape coincide with helices (383–396 and 402–415) previously observed in molecular dynamics (MD) simulations.
変異ランドスケープで観察された保存されたストレッチは、分子動力学 (MD) シミュレーションで以前に観察されたヘリックス (383~396 および 402~415) と一致しています。
In further support, we find structural prediction displays transient helices in this region, in the AlphaFold2 result spanning residues 400–410 (SI Appendix Fig. S3).
さらに、構造予測では、残基 400~410 にまたがる AlphaFold2 の結果で、この領域に一時的なヘリックスが表示されていることがわかりました (SI 付録図 S3)。
The relatively low confidence score is expected given the intrinsic disorder of the C-arm, but nonetheless suggests a propensity for transient helix formation.
C アームの固有の無秩序性を考えると、比較的低い信頼スコアが予想されますが、それでも一時的なヘリックス形成の傾向があることを示唆しています。
Structure predictions of segment Q390-A419 by other methods (I-TASSER and Phyre2) also suggest helical content, with the highest score for the segment 400–408.
他の方法 (I-TASSER および Phyre2) によるセグメント Q390-A419 の構造予測でもヘリックス含有量が示唆されており、セグメント 400~408 のスコアが最も高くなっています。
For the protected leucine-rich sequence 218–231, a potential role arises from its location within the linker region 210–246 found to be essential for RNA-mediated LLPS.
保護されたロイシンリッチ配列 218–231 については、RNA 媒介 LLPS に必須であることがわかっているリンカー領域 210–246 内の位置から潜在的な役割が生まれます。
The conserved island is also overlapping with the locus of a previously identified nuclear export signal 224–230.
保存された島は、以前に特定された核外輸送シグナル 224–230 の遺伝子座とも重なっています。
Incidentally, it overlaps with the peptide 222–230 that is a binder for HLA-A02:01 and immunogenic.
ちなみに、これは HLA-A02:01 の結合剤であり免疫原性があるペプチド 222~230 と重複しています。
Cubuk et al. reported transient helices in the leucine-rich region in MD simulations, proposed to provide oligomerization interfaces.
Cubuk らは、MD シミュレーションでロイシンに富む領域に一時的なヘリックスが存在することを報告し、オリゴマー化インターフェースを提供することを提案しました。
Using structure-prediction tools, we confirm the presence of a helical segment spanning residues 215–235 (SI Appendix Fig. S3), and, as further described below, and find evidence for its role in protein oligomerization and co-assembly with NA.
構造予測ツールを使用することで、残基 215~235 にまたがるらせんセグメントの存在を確認しました (SI 付録図 S3)。また、以下にさらに説明するように、タンパク質オリゴマー化および NA との共集合におけるその役割の証拠も見つかりました。
On the N-terminal half of the disordered linker, the SR-rich region 176–206 has been a locus of particular interest due to the cluster of charged residues and phosphorylation sites.
無秩序なリンカーの N 末端半分では、SR に富む領域 176~206 が、荷電残基とリン酸化部位のクラスターのため、特に興味深い場所となっています。
Their phosphorylation state is thought to regulate N-protein functions and interaction with the viral NSP3 protein and host proteins such as glycogen synthase kinase-3, CDK-1, and 14-3-3 proteins.
これらのリン酸化状態は、N タンパク質の機能と、ウイルス NSP3 タンパク質およびグリコーゲン合成酵素キナーゼ 3、CDK-1、14-3-3 タンパク質などの宿主タンパク質との相互作用を制御すると考えられている。
Focusing on the 14 serine residues in this stretch, the mutational landscape shows that, with the exception of completely conserved S176, all other phosphorylation sites can be substituted, and new ones can be introduced.
この範囲の 14 個のセリン残基に焦点を当てると、変異の状況から、完全に保存されている S176 を除き、他のすべてのリン酸化部位は置換可能であり、新しい部位が導入される可能性があることが分かります。
Overall, 14.3% of all sequences exhibit changes in the serine pattern; however, there is a significant anti-correlation to maintain a total number of 13 or more serines in this region (Table S2).
全体として、すべての配列の 14.3% でセリンパターンの変化が見られますが、この領域では合計 13 個以上のセリンを維持するために大きな逆相関があります (表 S2)。
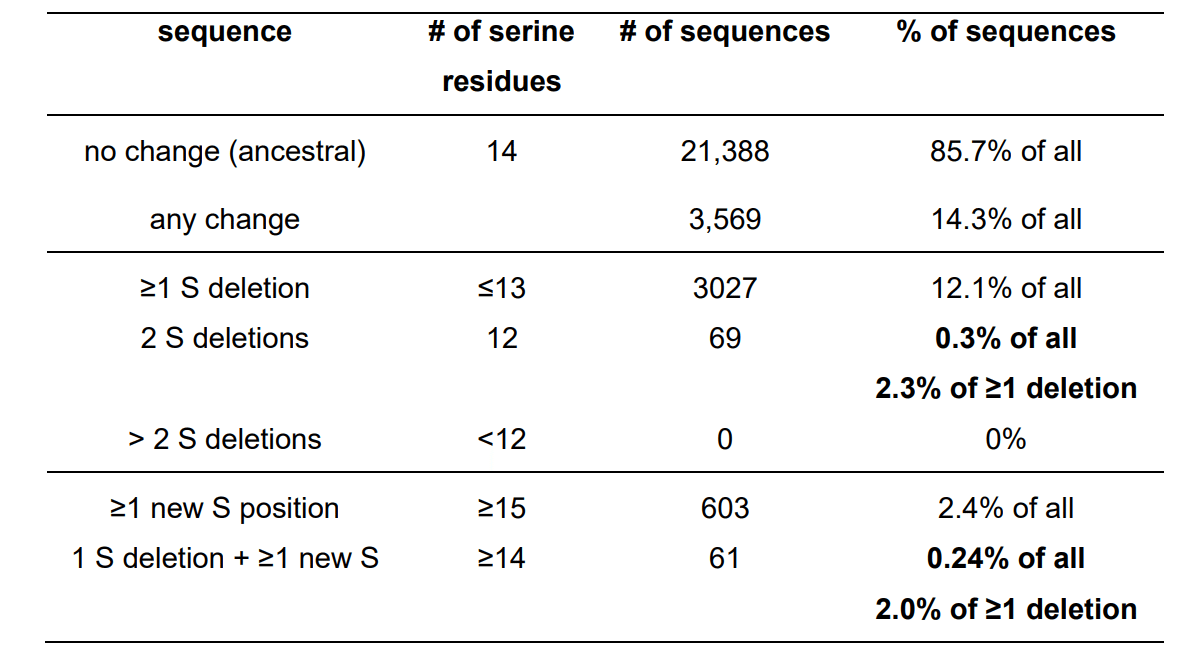
Thus, it appears that except for S176, there is redundancy and flexibility in the phosphorylation sites, but with a constraint to maintain their local density.
したがって、S176 を除いて、リン酸化部位には冗長性と柔軟性がありますが、局所的な密度を維持するという制約があるようです。
Within this SR-rich region, the R203 mutation was noted early in the pandemic and remained common to all variants of interest.
このSRに富む領域では、パンデミックの初期にR203変異が注目され、関心のあるすべての変異体に共通していました。
The earlier R203K/G204R was shown experimentally to enhance the ability of N-protein to form condensates, and R203M – prevalent in the Delta variant – was recently reported to enhance viral replication.
以前の R203K/G204R は、N タンパク質の凝縮体形成能力を高めることが実験的に示されており、デルタ変異体によく見られる R203M は、最近、ウイルスの複製を高めることが報告されました。
The most recent mutation G215C is located in the linker between the SR-rich and leucine rich regions.
最も最近の変異 G215C は、SR に富む領域とロイシンに富む領域の間のリンカーにあります。
It has arisen in the Delta 21J clade during the last several months.
これは、過去数か月間に Delta 21J 系統で発生しました。
Accompanied solely by mutations in ORF1ab and ORF7b, it quickly outcompeted any variants not containing G215C to assume worldwide dominance (Fig. 1b).
ORF1ab と ORF7b の変異のみを伴い、G215C を含まない変異体をすぐに打ち負かし、世界中で優位に立っています (図 1b)。
As of November 29, 2021, 49.6 % of sequences in the data base contain the G215C mutation, but these describe only 34.9% of unique sequences (8,720), consistent with the shorter period of time since the 21J clade has emerged.
2021 年 11 月 29 日現在、データベース内の配列の 49.6 % に G215C 変異が含まれていますが、これらは固有の配列の 34.9 % (8,720) のみを説明しています。 これは、21J 系統が出現してからの期間が短いことと一致しています。
Nevertheless, when examining to what extent these have already expanded to reproduce the mutational landscape, we find 97.2% of instances of N mutations occur in positions that show mutations in both 21J and non-21J clades (Fig. 3c, see SI Appendix Fig. S4 for a detailed list).
それでも、これらがどの程度拡大して変異ランドスケープを再現しているかを調べると、N 変異の 97.2% が 21J および非 21J 系統の両方で変異を示す位置で発生していることがわかります (図 3c、詳細なリストについては SI 付録の図 S4 を参照)。
Incomplete overlap can be discerned mostly among rare substitutions and in highly disordered regions.
不完全な重複は、主にまれな置換と高度に無秩序な領域で識別できます。
Quantitatively, 14.0% of distinct mutations in non-21J (pre-Delta) species have not yet been observed in the 21J clade, potentially due to still incomplete coverage.
量的には、非 21J (デルタ以前) 種の明確な変異の 14.0% が 21J 系統ではまだ観察されていません。これは、カバー範囲がまだ不完全である可能性が考えられます。
Interestingly, however, 21.8% of distinct substitutions in the 21J clade were not previously observed in non-21J clades, suggesting evolution of biophysical properties (Fig. 1c).
しかし、興味深いことに、21J 系統の明確な置換の 21.8% は、非 21J 系統ではこれまで観察されておらず、生物物理学的特性の進化を示唆しています (図 1c)。
N-protein mutants display altered secondary structure and assembly properties
Nタンパク質変異体は二次構造と組み立て特性が変化
To study the impact of the mutation on structure and assembly function of N-protein, we compare the biophysical properties of select N-protein mutants derived from the Delta variant with those of the ancestral Wuhan-Hu-1 N-protein (Nref) in vitro.
変異がNタンパク質の構造とアセンブリ機能に与える影響を研究するため、Delta変異体から派生した特定のNタンパク質変異体の生物物理学的特性と、祖先のWuhan-Hu-1 Nタンパク質(Nref)の生物物理学的特性をin vitroで比較しました。
First, we examined the coarse-grained size and shape using sedimentation velocity analytical ultracentrifugation (SV) and dynamic light scattering (DLS).
まず、沈降速度分析超遠心分離(SV)と動的光散乱(DLS)を使用して、粗粒度のサイズと形状を調べました。
As shown previously, when expressed in E. coli and purified to remove NA, Nref forms non-covalent 4.1 S dimers with a Stokes radius of 5.9 nm.
以前に示したように、大腸菌で発現し、NA を除去するために精製すると、Nref はストークス半径 5.9 nm の非共有結合 4.1 S 二量体を形成します。
Its translational frictional ratio of 1.82 indicates a highly extended hydrodynamic shape as a result of significant disorder.
その並進摩擦比 1.82 は、著しい無秩序の結果として、非常に拡張された流体力学的形状を示しています。
The dimers are linked tightly at the dimerization interface in the CTD with KD < 10 nM, but show only ultra-weak further self-association (KD = 760 μM).
二量体は、CTD の二量体化インターフェースで KD < 10 nM でしっかりと結合していますが、さらに非常に弱い自己会合 (KD = 760 μM) しか示しません。
An overlay of sedimentation coefficient distributions of the N:D63G and N:G215C mutants, as well as the quadruple N:D63G,R203M,G215C,D377Y mutant reflecting the full set of canonical mutations in 21J Delta variant is shown in Fig. 4.
図 4 に、N:D63G および N:G215C 変異体、および 21J Delta 変異体の標準的な変異の完全なセットを反映した 4 重 N:D63G、R203M、G215C、D377Y 変異体の沈降係数分布の重ね合わせを示します。
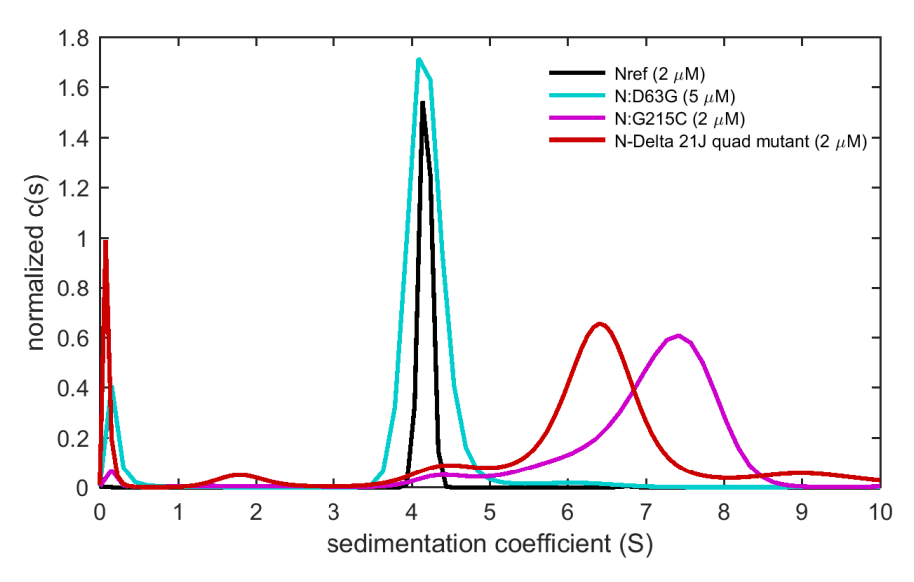
While N:D63G sediments similar to Nref, indicating no change in size, shape, or self-association properties, both N:G215C and the quadruple mutant sediment much faster, at a rate that demonstrates the formation of tetramers at low micromolar concentrations.
N:D63G は Nref と同様に沈降し、サイズ、形状、または自己会合特性に変化がないことを示していますが、N:G215C と 4 重変異体はどちらもはるかに速く沈降し、低マイクロモル濃度でテトラマーの形成を示す速度です。
To comprehensively examine the altered state we focus on N:G215C, in light of its unique epidemiological impact, and to enable clear structural attribution of changes to a single residue substitution.
変化した状態を包括的に調査するために、その独特の疫学的影響を考慮して N:G215C に焦点を当て、変化が単一の残基置換に構造的に明確に帰属できるようにします。
Compared to the ancestral Nref, N:G215C sediments much faster at 7.3 S (Fig. 5a), with a lower hydrodynamic frictional ratio of 1.58, in a relatively more compact, tetrameric solution state with a Stokes radius of 7.09 nm (Fig. 5c).
先祖の Nref と比較すると、N:G215C は 7.3 S ではるかに速く沈降し (図 5a)、流体摩擦比は 1.58 と低く、ストークス半径が 7.09 nm の比較的コンパクトな四量体溶液状態になります (図 5c)。
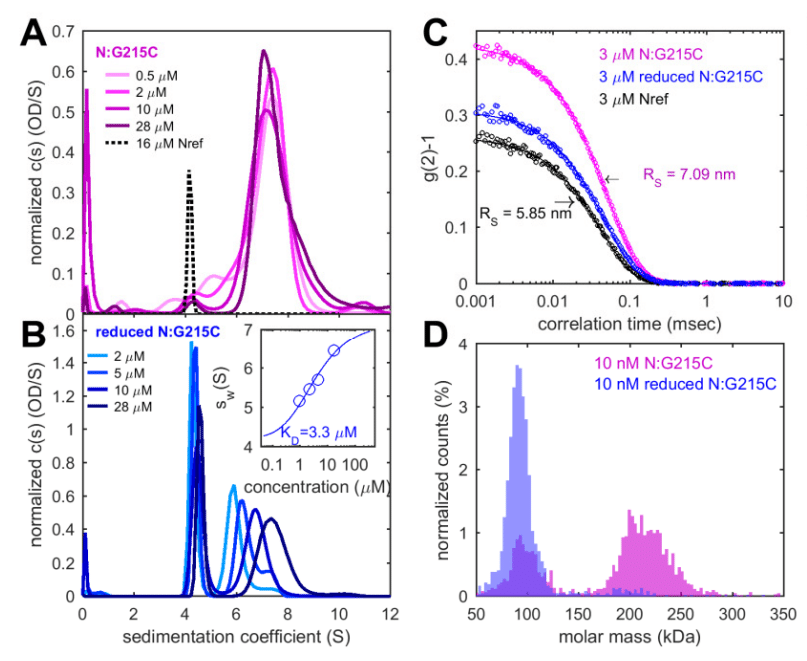
Electrospray mass spectrometry (ESI-MS) shows an intact mass of 93,792 Da, consistent with the expected value for a His-tagged dimer.
エレクトロスプレー質量分析 (ESI-MS) では、質量は 93,792 Da のままで、His タグ付き二量体の予想値と一致しています。
Furthermore, LC-MS/MS of tryptic digests shows a peptide consistent with a species composed of two tryptic peptides crosslinked by disulfide bond at 215C.
さらに、トリプシン消化物の LC-MS/MS では、215C でジスルフィド結合によって架橋された 2 つのトリプシンペプチドからなる種と一致するペプチドが示されました。
Finally, we used mass photometry (MP) to obtain an independent direct measurement of the molecular weight distribution of N:G215C in solution (Fig. 5d).
最後に、質量測定 (MP) を使用して、溶液中の N:G215C の分子量分布の独立した直接測定を取得しました (図 5d)。
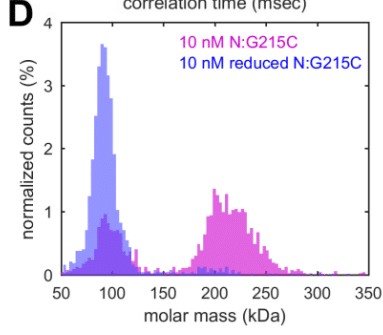
Different from ESI-MS, this method leaves the non-covalent dimerization at the CTD intact.
ESI-MS とは異なり、この方法では CTD での非共有結合二量化がそのまま残ります。
Accordingly, a majority peak can be discerned approximately at the tetramer mass.
したがって、大部分のピークはほぼテトラマー質量で識別できます。
In summary, N:G215C forms a tightly bound, compact tetramer via disulfide crosslinks of non-covalent dimers.
要約すると、N:G215C は、非共有結合二量体のジスルフィド架橋を介して、しっかりと結合したコンパクトなテトラマーを形成します。
When disrupting disulfide bonds in reducing conditions, as expected, the intact mass obtained from ESI-MS was 46,897 Da, consistent with the mass of the monomer.
還元条件下でジスルフィド結合を切断すると、予想通り、ESI-MS から得られた完全な質量は 46,897 Da で、モノマーの質量と一致しました。
By MP, the mass distribution exhibits a peak at the dimer molecular weight (Fig. 5d), consistent with significant non-covalent CTD dimerization similar to Nref.
MP では、質量分布は二量体分子量でピークを示し (図 5d)、Nref に類似した顕著な非共有結合 CTD 二量体化と一致しています。
However, at the much higher μM concentrations in SV, reduced N:G215C shows much different behavior (Fig. 5b): In addition to a major peak at 4.1 S for the dimer, a faster sedimenting population with distinct concentration dependence can be discerned.
しかし、SV のはるかに高い μM 濃度では、還元型 N:G215C は大きく異なる挙動を示します (図 5b):4.1 S での二量体の主要ピークに加えて、明確な濃度依存性を持つより速い沈降集団が識別できます。
This is characteristic of further self-association in rapid association/dissociation exchange.
これは、急速な会合/解離交換におけるさらなる自己会合の特徴です。
An isotherm of weigh-average s-values can be modeled as a dimer-tetramer association step with a best-fit dimer KD of 3.3 μM (Fig. 5b inset).
重量平均 s 値の等温線は、最適な二量体 KD が 3.3 μM である二量体-四量体会合ステップとしてモデル化できます (図 5b 挿入図)。
This is ∼200-fold stronger than previously measured for Nref.
これは、Nref について以前に測定されたものより約 200 倍強力です。
Thus, the G215C mutation induces conformational alterations that create, or significantly enhance, a non-covalent dimer-dimer protein interaction interface outside the CTD, even in the absence of covalent disulfide bonds.
したがって、G215C 変異は、共有結合ジスルフィド結合がない場合でも、CTD の外側に非共有結合二量体-二量体タンパク質相互作用界面を作成するか、大幅に強化する構造変化を引き起こします。
The possibility of covalent dimerization of CTD-linked dimers poses a question regarding the quaternary structure, since multivalent dimers could potentially self-assemble into wide range of higher-order oligomers (Fig. 5f).
CTD 結合二量体の共有結合二量体化の可能性は、多価二量体が潜在的に幅広い高次オリゴマーに自己組織化する可能性があるため、四次構造に関する疑問を提起します (図 5f)。
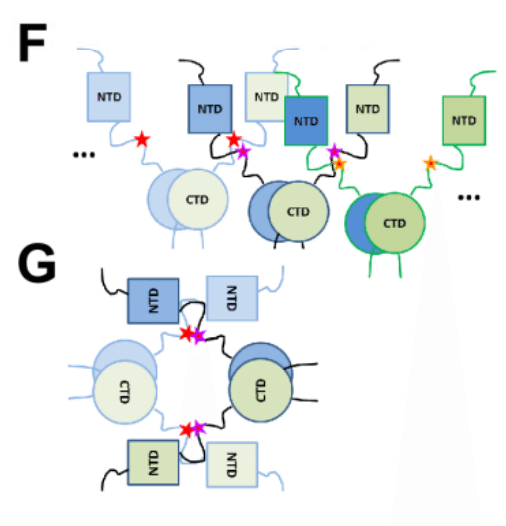
Such structures would be detected with high sensitivity both in SV and DLS but are virtually absent in our data.
このような構造は SV と DLS の両方で高感度に検出されますが、私たちのデータには事実上存在しません。
Instead, as supported by reduction and re-oxidation experiments (SI Appendix Fig. S5), measurement of free sulfhydryls (SI Appendix Fig. S6), and the measured compact hydrodynamic shape (Fig. 5a, c), we propose a more compact configuration with two intra-tetramer crosslinks (Fig. 5g).
代わりに、還元および再酸化実験 (SI 付録図 S5)、遊離スルフヒドリルの測定 (SI 付録図 S6)、および測定されたコンパクトな流体力学的形状 (図 5a、c) によって裏付けられているように、2 つのテトラマー内架橋を持つよりコンパクトな構成を提案します (図 5g)。
Unfortunately, published cryo-ET structures do not yet allow unambiguous structural assignment of N-protein configuration in the ribonucleoprotein particles.
残念ながら、公開されているクライオ ET 構造では、リボ核タンパク質粒子内の N タンパク質構成の明確な構造割り当てはまだできません。
To complement the study of coarse-grained aspects of N-protein size and shape, we examined the secondary structure content of N:G215C by circular dichroism spectroscopy (CD).
N タンパク質のサイズと形状の粗視化に関する研究を補完するために、円二色性分光法 (CD) によって N:G215C の二次構造含有量を調べました。
Whereas the spectrum of Nref is dominated by a large negative ellipticity at 200 nm that is characteristic for disordered chains, N:G215C shows much reduced negative 200 nm signal and instead stronger ellipticity in the range 220-230 nm typical for helical structures (Fig. 5e).
Nref のスペクトルは、無秩序な鎖に特徴的な 200 nm での大きな負の楕円率によって支配されていますが、N:G215C は、負の 200 nm 信号が大幅に減少し、代わりにらせん構造に典型的な 220~230 nm の範囲でより強い楕円率を示しています (図 5e)。
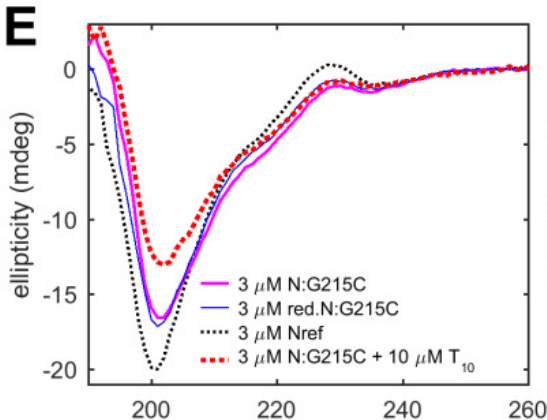
Such diminished disorder is consistent with the more compact hydrodynamic shape of N:G215C.
このような無秩序性の減少は、N:G215C のよりコンパクトな流体力学的形状と一致しています。
Virtually identical spectra were obtained in reducing conditions, and across a concentration range populating different fractions of dimers and tetramers.
還元条件では、また、二量体と四量体の異なる割合を占める濃度範囲全体で、実質的に同一のスペクトルが得られました。
A key step in assembly is the interaction between NA and N-protein.
組み立ての重要なステップは、NA と N タンパク質の相互作用です。
We previously probed consequences of NA binding on N-protein interactions by studying N-protein liganded with short oligonucleotides.
我々は以前、短いオリゴヌクレオチドを結合させた N タンパク質を研究することで、NA 結合が N タンパク質相互作用に与える影響を調査した。
Up to a decanucleotide T10, a length that spans the binding grove of the NTD, we observed similar binding affinities of NA for N:G215C as previously determined for Nref (SI Appendix Fig. S7).
NTD の結合溝にまたがる長さであるデカヌクレオチド T10 まで、N:G215C に対する NA の結合親和性は、以前に Nref に対して決定されたものと同様であることが観察されました (SI 付録図 S7)。
Likewise, virtually unaltered is a reduction in disorder by CD spectroscopy when liganded by T10 (Fig. 5e), as well as a NA binding-related shift to greater thermal stability of the folded domains observed by differential scanning fluorometry (SI Appendix Fig. S8).
同様に、T10 でリガンド結合した場合の CD 分光法による無秩序性の減少 (図 5e) や、示差走査蛍光測定法で観察された折り畳まれたドメインの NA 結合関連の熱安定性の向上 (SI 付録図 S8) も、実質的に変化していません。
Apparently, elementary features of NA binding are not substantially affected by the G215C mutation.
明らかに、NA 結合の基本的特徴は、G215C 変異によって実質的に影響を受けません。
A much different picture arises when binding the longer oligonucleotides T20.
より長いオリゴヌクレオチド T20 を結合すると、まったく異なる図が浮かび上がります。
These spatially extend beyond a single NTD domain and can bridge between two N-protein dimers.
これらは単一の NTD ドメインを超えて空間的に広がり、2 つの N タンパク質二量体の間を橋渡しすることができます。
For Nref, they promote formation of tetramers and higher oligomers at low micromolar protein concentrations.
Nref の場合、低マイクロモル濃度のタンパク質でテトラマーおよびより高次のオリゴマーの形成を促進します。
In the case of N:G215C, we find this co-assembly significantly augmented.
N:G215C の場合、この共集合が大幅に強化されていることがわかります。
While this may be expected for disulfide-linked N:G215C tetramer, even in the reduced conditions N:G215C exhibits significantly stronger hetero-oligomerization (Fig. 5h).
これはジスルフィド結合した N:G215C テトラマーでは予想されることですが、還元条件でも N:G215C は大幅に強いヘテロオリゴマー化を示します (図 5h)。
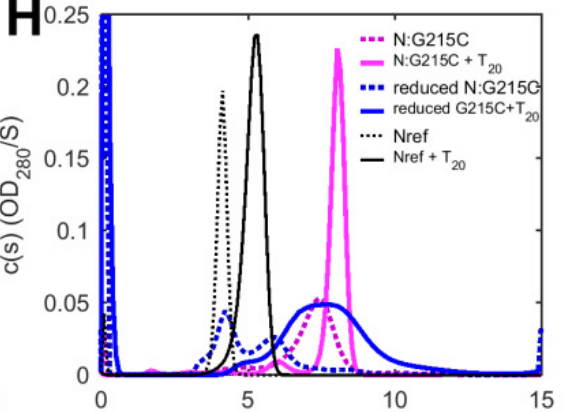
This shows cooperativity or an avidity advantage of N:G215C in the earliest steps of assembly with NA, presumably due to its ability to constitutively tetramerize.
これは、NA との集合の最も初期の段階で N:G215C が協同性または親和性優位性を持っていることを示しています。これは、おそらく N:G215C が構成的にテトラマー化する能力を持っているためです。
Finally, we compare aspects of higher-order assembly and LLPS.
最後に、高次アセンブリと LLPS の側面を比較します。
LLPS depends on highly multi-valent, weak interactions, such as transient aromatic side-chain and backbone interactions of disordered chains.
LLPS は、不規則な鎖の一時的な芳香族側鎖とバックボーンの相互作用など、非常に多価で弱い相互作用に依存します。
These can be expected to differ from protein/NA and protein-protein interactions that stabilize the discrete oligomeric co-assemblies observed above, including the tetramerization property augmented by the G215C mutation.
これらは、G215C 変異によって増強された四量体化特性を含む、上で観察された個別のオリゴマー共集合体を安定化するタンパク質/NA およびタンパク質-タンパク質相互作用とは異なることが予想されます。
LLPS of Nref can be induced at higher temperature and by nucleic acid binding.
Nref の LLPS は、高温および核酸結合によって誘発される可能性があります。
It is preceded by the formation of ∼0.1 – 1 μm sized clusters.
その前に、約 0.1 – 1 μm サイズのクラスターが形成されます。
This is accompanied by structural transitions by CD, which we similarly observe for N:G215C (SI Appendix Fig. S9).
これには CD による構造遷移が伴い、これは N:G215C でも同様に観察されます (SI 付録図 S9)。
However, N:G215C exhibits much steeper transitions, and at a lower transition temperature, as may be discerned from the temperature-dependent particle size in DLS (Fig. 5i).
ただし、DLSの温度依存粒子サイズからわかるように、N:G215Cははるかに急峻な遷移を示し、遷移温度も低くなります(図5i)。
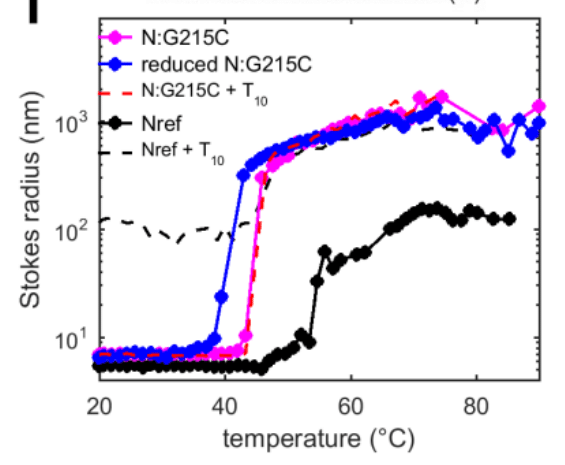
Interestingly, while for Nref a lower phase transition temperature is achieved in the presence of T10, the same transition temperature is observed for N:G215C already without any NA, and addition of T10 to N:G215C does not lead to a further shift.
興味深いことに、Nref では T10 の存在下でより低い相転移温度が達成されるのに対し、NA が存在しない N:G215C ではすでに同じ転移温度が観察されており、N:G215C に T10 を追加してもそれ以上のシフトは起こりません。
Removal of disulfide bonds in reducing conditions further lowered the phase transition temperature.
還元条件下でのジスルフィド結合の除去により、相転移温度はさらに低下しました。
These results indicate more cooperative assembly with lower energy barrier.
これらの結果は、エネルギー障壁が低い、より協調的なアセンブリを示しています。
The largest objects in the co-assembly process of N-protein and NA in vitro are droplets from LLPS that are visible in light microscopy.
Nタンパク質とNAのin vitro共集合プロセスにおける最大の物体は、光学顕微鏡で観察できるLLPSからの液滴です。
At 20°C (i.e. below the transition temperature) Nref and N:G215C only exhibited small differences, with slightly larger droplets observed for reduced N:G215C compared to Nref (SI Appendix Fig. S10).
20°C(つまり、遷移温度未満)では、NrefとN:G215Cはわずかな違いしか示さず、還元型N:G215CではNrefと比較してわずかに大きい液滴が観察されました(SI付録図S10)。
Structural Basis of Protected Islands in Disordered Regions and Effects of the G215C mutation
無秩序地域における保護された島の構造的基礎とG215C変異の影響
While several NMR and crystallographic structures are available for the NTD and CTD domains, the features of the structure and dynamics of the disordered linker and arms are less well understood.
NTD および CTD ドメインにはいくつかの NMR 構造と結晶構造がありますが、不規則なリンカーとアームの構造とダイナミクスの特徴はあまりよく理解されていません。
The mutationally protected sequence islands in the C-arm at 390–394 and 403–408 and the central linker region 218–231 coincide with transient helices previously revealed in MD simulations.
390~394 と 403~408 の C アームの変異保護された配列アイランドと中央のリンカー領域 218~231 は、以前に MD シミュレーションで明らかにされた一時的なヘリックスと一致しています。
As described above, helices in these segments are also found with different structure prediction methods (Fig. 6a and SI Appendix Fig. S3).
前述のように、これらのセグメントのヘリックスも、さまざまな構造予測方法で検出されています (図 6a および SI 付録の図 S3)。
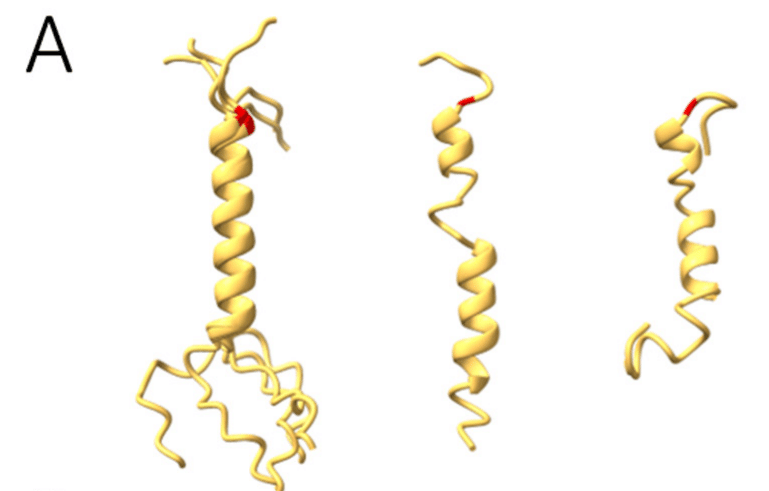
To assess the impact of the G215C mutation, we studied the disordered stretch 210–246 in the central linker containing the leucine-rich region.
G215C 変異の影響を評価するために、ロイシンに富む領域を含む中央リンカーの無秩序なストレッチ 210~246 を調べました。
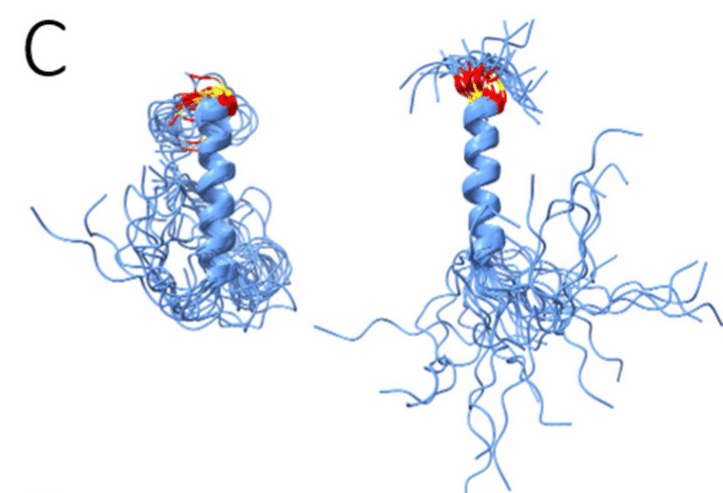
Although structure prediction is generally poor in the disordered segments, all the models show varying degrees of α-helical structure in the sequence of interest, with G215 at the stem of the helix.
構造予測は不規則なセグメントでは一般的に不十分ですが、すべてのモデルは、関心のある配列にさまざまな程度のαヘリックス構造を示しており、G215はヘリックスの幹にあります。
Highest reliability is obtained in the 222– 234 stretch (Fig. 6a).
222〜234ストレッチで最も高い信頼性が得られています(図6a)。
The top-ranked AlphaFold2 model was used here as the initial structure for MD calculations.
最上位のAlphaFold2モデルは、ここではMD計算の初期構造として使用されました。
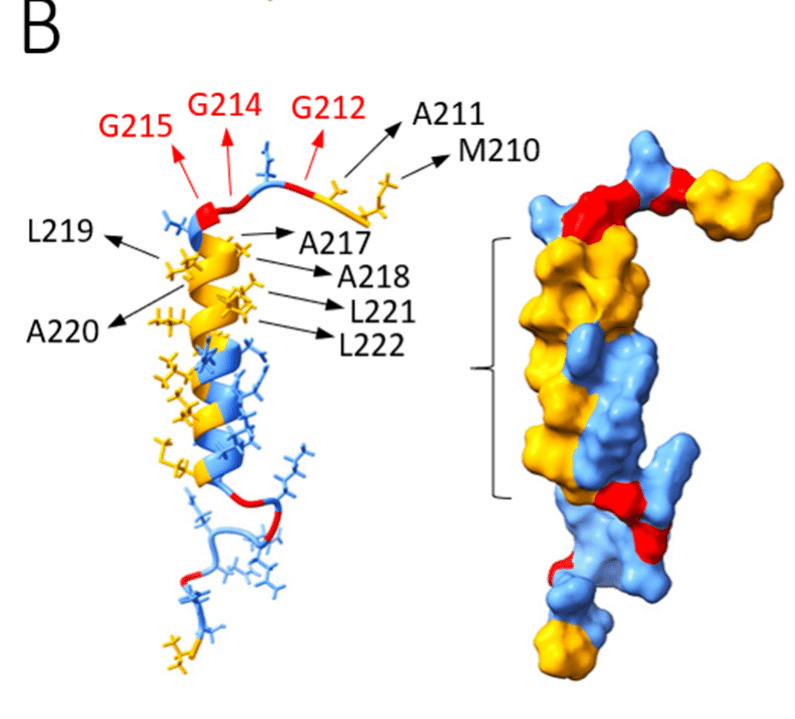
Key residues of the helix can be discerned in Fig. 6b, which highlights six flanking Gly residues conferring structural flexibility, and a bed of hydrophobic residues that stabilize the helix and may serve as a protein-protein interaction interface.
ヘリックスの主な残基は図 6b で識別できます。図では、構造的柔軟性を与える 6 つの隣接する Gly 残基と、ヘリックスを安定化し、タンパク質間相互作用インターフェースとして機能する可能性のある疎水性残基の層が強調されています。
Mutation of G215 by C215 results in redirection and reduced flexibility of the N-term of the helix (Fig. 6c).
G215 を C215 に変異させると、らせんの N 末端の方向転換と柔軟性の低下が起こります (図 6c)。
These structural and dynamic changes of the mutant originate both in the higher flexibility of Gly relative to Cys and in the stabilization of the Cys sidechain through persistent hydrophobic interactions with A218 and L219 (Fig. 6d).
変異体のこれらの構造的および動的変化は、Cys に対する Gly の柔軟性の高さと、A218 および L219 との持続的な疎水性相互作用による Cys 側鎖の安定化の両方に起因しています (図 6d)。
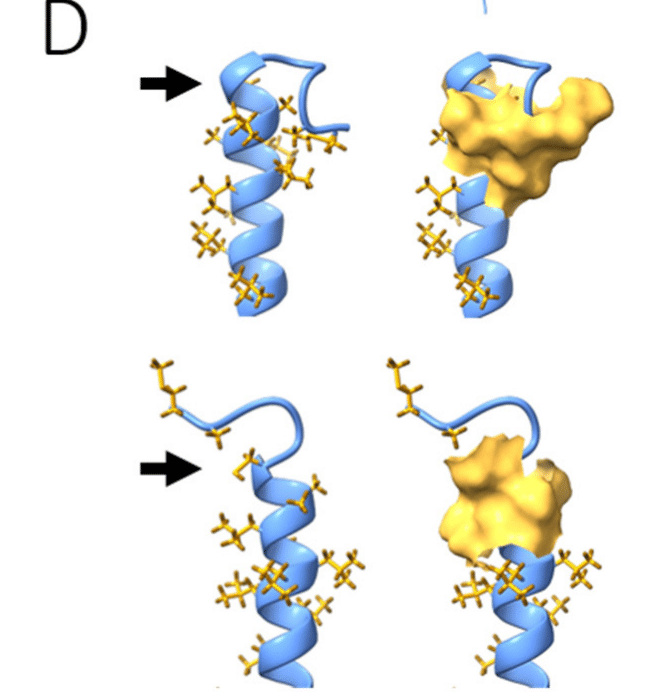
This results in a more open configuration that appears better poised for helix-helix and thiol interactions.
これにより、ヘリックス間およびチオール相互作用に適した、よりオープンな構成になります。
This is consistent with the experimentally observed enhanced dimer-dimer interactions of the N:G215C under reduced conditions and the possibility of forming disulfide bonds across different protomers.
これは、還元条件下でのN:G215Cの実験的に観察された強化された二量体間相互作用と、異なるプロトマー間でジスルフィド結合を形成する可能性と一致しています。
Discussion
The worldwide sequencing effort has led to the assembly of an unprecedented database of viable SARS-CoV-2 variants, alone for N-protein describing ∼25,000 different species as of November 2021.
世界的な配列解析の取り組みにより、2021年11月現在、Nタンパク質だけでも約25,000種の異なる種を記述する、前例のないSARS-CoV-2の生存可能な変異体のデータベースが構築されました。
In the present work we have combined an analysis of the amino acid variability of SARS-CoV-2 N-protein with biophysical experiments of select mutants, and found remarkable plasticity on all levels of organization.
本研究では、SARS-CoV-2 Nタンパク質のアミノ酸変異性の分析と選択された変異体の生物物理学的実験を組み合わせ、組織のすべてのレベルで顕著な可塑性を発見しました。
Plasticity has been hypothesized to be a unique feature of RNA virus proteins, which have more loosely packed cores and an abundance of intrinsically disordered regions that confer high degrees of flexibility for adaptation and tolerance to mutations.
可塑性は、RNAウイルスタンパク質のユニークな特徴であると仮定されてきた。RNAウイルスタンパク質は、より緩く詰まったコアと、突然変異に対する適応と耐性のための高度な柔軟性を与える本質的に無秩序な領域を豊富に持っている。
In the present case of the SARS-CoV-2 N-protein, more than 86% of positions in the amino acid sequence are subject to variation, on average by 3-4 different amino acids, about half of which score low on a physicochemical similarity scale.
SARS-CoV-2 Nタンパク質の今回のケースでは、アミノ酸配列の位置の86%以上が、平均して3~4種類のアミノ酸の変化を受けており、そのうち約半分は物理化学的類似性スケールで低いスコアとなっています。
We observed substitutions at many positions thought to be critical due to their conservation across related coronaviruses, or their constitution of phosphorylation and protein interaction sites, are found to be viable.
関連するコロナウイルス間での保存性、またはリン酸化とタンパク質相互作用部位の構成により重要と考えられている多くの位置での置換が、実行可能であることがわかりました。
Our data show that the single mutation G215C causes significantly altered secondary structure; gross differences in hydrodynamic shape indicate altered subunit arrangements; and strongly enhanced protein-protein interactions modulate the formation of quaternary structure.
私たちのデータは、単一の変異G215Cが二次構造の大幅な変化を引き起こし、流体力学的形状の大きな違いがサブユニット配置の変化を示し、タンパク質間相互作用の大幅な強化が四次構造の形成を調節することを示しています。
More extended studies are required to elucidate the expected impact of mutations in N-protein, for example, on host protein interactions, interactions with other viral proteins, and on phase boundaries for condensation and nucleic acid coassembly.
Nタンパク質の変異が、例えば宿主タンパク質相互作用、他のウイルスタンパク質との相互作用、凝縮と核酸共集合の相境界に及ぼす予想される影響を明らかにするには、さらに広範な研究が必要です。
This plasticity poses significant challenges to identify the essential functions and mechanisms that may serve as therapeutic targets in N-protein.
この可塑性は、Nタンパク質の治療標的となる可能性のある重要な機能とメカニズムを特定する上で大きな課題を提起しています。
Earlier examinations of emerging mutations in N proteins, going back to June 2020, were necessarily more limited in scope, and while sufficient to examine hot spots and identify key replacements such as R203K/G204R, it was not yet possible to draw conclusions from a survey of the entire mutational landscape.
2020 年 6 月まで遡る N タンパク質の新たな変異に関する以前の調査は、必然的に範囲がより限定されており、ホットスポットを調査して R203K/G204R などの主要な置換を特定するには十分でしたが、変異ランドスケープ全体の調査から結論を導き出すことはまだできませんでした。
Due to the orders of magnitude larger coverage that has become available through the global genomic epidemiology efforts since then, we believe that the observed data now approaches the limits of possible mutations for functioning N-protein, and therefore reflects its biophysical properties.
それ以降、世界的なゲノム疫学の取り組みを通じて利用できるようになったカバレッジは桁違いに大きくなったため、観察されたデータは現在、機能する N タンパク質の可能な変異の限界に近づいており、したがってその生物物理学的特性を反映していると考えています。
The study of the constraints in the mutational landscape appears very useful to complement structural biophysical tools, particularly to examine intrinsically disordered regions.
変異ランドスケープにおける制約の研究は、構造生物物理学的ツールを補完するのに非常に有用であるように思われ、特に本質的に無秩序な領域を調べるのに有用である。
These generally are permissive for a wide range of amino acid substitutions and, in fact, harbor three of the four persistent N-protein mutations characteristic of the Delta variant, and all of the Omicron mutations in N-protein.
これらは一般に、広範囲のアミノ酸置換を許容し、実際、デルタ変異体に特徴的な 4 つの持続的な N タンパク質変異のうち 3 つと、N タンパク質のオミクロン変異のすべてを抱えている。
However, the mutational landscape of N-protein reveals several islands within these regions that are highly protected and therefore appear to have critical functions.
しかし、N タンパク質の変異ランドスケープは、これらの領域内に高度に保護されており、したがって重要な機能を持つと思われるいくつかの島を明らかにしている。
One of these is in the central linker adjacent to the G215 position, where the G215C mutation has quickly outcompeted all other variants in 2021 after its appearance alongside only ORF1ab and ORF7b mutations in the Delta variant.
これらのうちの1つは、G215位置に隣接する中央リンカーにあり、G215C変異は、Delta変異体でORF1abおよびORF7b変異と並んで出現した後、2021年に他のすべての変異体を急速に上回りました。
NMR and molecular dynamics studies have reported distinct α-helical propensity flanking position 215 in the SR-rich region and in the leucine-rich region.
NMRおよび分子動力学研究では、SRに富む領域とロイシンに富む領域で、位置215に隣接する明確なαヘリックス傾向が報告されています。
The latter was proposed by Cubuk et al. to provide interfaces for oligomerization.
後者は、Cubuk らによってオリゴマー化のためのインターフェースを提供するために提案されました。
Most recently, NMR experiments showed formation of an α-helix 219-230 when in complex with nsp3a, which binds to N-protein competitively with RNA.
最近では、NMR 実験により、nsp3a と複合体を形成すると 219-230 の α ヘリックスが形成されることが示されました。 nsp3aは RNA と競合的に N タンパク質に結合します。
In the present work, we found this region highly protected against mutation.
本研究では、この領域が変異に対して高度に保護されていることを発見しました。
We propose that these helices may be essential for higher-order assembly of N-protein and are either stabilized or exposed in the emerging G215C mutant.
これらのヘリックスは、N タンパク質の高次アセンブリに不可欠であり、出現した G215C 変異体では安定化または露出していると考えられます。
Indeed, our simulations show a stable helix spanning residues G215-S235 exhibiting an extended hydrophobic surface on one side.
実際、シミュレーションでは、残基 G215-S235 にまたがる安定したヘリックスが、片側に拡張された疎水性表面を示しています。
Mutation to C215 repositions the downstream sequence relative to the helix, which renders the hydrophobic surface more accessible for protein-protein interactions.
C215 への変異により、ヘリックスに対する下流の配列の位置が変更され、疎水性表面がタンパク質間相互作用に対してよりアクセスしやすくなります。
The potential introduction of disulfide bonds in the N:G215C mutant stabilizing dimer-dimer crosslinks in the linker region would constitute an even more drastic change in the assembly pathway.
N:G215C 変異体にジスルフィド結合が導入され、リンカー領域で二量体間架橋が安定化すると、組み立て経路にさらに劇的な変化が生じる可能性があります。
It is uncertain, however, whether these disulfide bonds are formed in vivo.
ただし、これらのジスルフィド結合が生体内で形成されるかどうかは不明である。
Disulfide bonds are absent in nucleocapsid proteins of related betacoronaviruses, but assist oligomerization of nucleocapsid proteins of other RNA viruses at least transiently.
ジスルフィド結合は、関連ベータコロナウイルスのヌクレオカプシドタンパク質には存在しないが、少なくとも一時的には他のRNAウイルスのヌクレオカプシドタンパク質のオリゴマー化を助ける。
However, even without disulfide bonds, we found a 200-fold enhancement of dimer-dimer self-association in N:G215C, accompanied by augmented co-assembly with NA, which we hypothesize profoundly alters the co-assembly kinetics of ribonucleoprotein particles.
しかし、ジスルフィド結合がない場合でも、N:G215C では二量体間の自己会合が 200 倍に増加し、NA との共集合が増強されていることが分かりました。このことは、リボ核タンパク質粒子の共集合動態を大きく変えると私たちは考えています。
This may contribute to the clinical phenotype of strongly increased viral load and infectivity of the Delta variant, and the overwhelming dominance of the 21J Delta clade relative to the 21I Delta clade despite the lack of additional changes in the spike protein.
これが、デルタ変異体のウイルス量と感染力が大幅に増加した臨床表現型、およびスパイクタンパク質にさらなる変化がないにもかかわらず 21I デルタ クレードに対する 21J デルタ クレードの圧倒的優位性に寄与している可能性があります。
After writing of this manuscript, the 21J Delta variant of SARS-CoV-2 has been replaced by the Omicron variant as the worldwide dominant variant.
この原稿の執筆後、SARS-CoV-2 の 21J Delta 変異体は、世界的に優勢な変異体として Omicron 変異体に取って代わられました。
Interestingly, the latter has none of the N-protein mutations defining for the Delta variant, but it exhibits deletions and new mutations that may impact viral fitness, in addition to the characteristic spike protein mutations.
興味深いことに、後者は Delta 変異体を特徴付ける N タンパク質変異をまったく持たず、特徴的なスパイクタンパク質変異に加えて、ウイルスの適応度に影響を与える可能性のある欠失と新しい変異を示しています。
While this development does not impact our conclusions on the mutational landscape of N-protein and its relation to its biophysical properties and assembly functions, it will be interesting to examine to which extent Omicron will explore, on a global population level, a similar N-protein sequence space as Delta and prior variants.
この展開は、N タンパク質の変異ランドスケープとその生物物理学的特性およびアセンブリ機能との関係に関する結論に影響を与えませんが、Omicron が世界人口レベルで Delta および以前の変異体と同様の N タンパク質配列空間をどの程度探索するかを調べることは興味深いでしょう。
以下省略。
この記事が気に入ったらサポートをしてみませんか?