BA.2.86の詳細な変異スキャンとKP.3変異体のエピスタシス的出現

Deep mutational scanning of SARS-CoV-2 Omicron BA.2.86 and epistatic emergence of the KP.3 variant
SARS-CoV-2 Omicron BA.2.86の詳細な変異スキャンとKP.3変異体のエピスタシス的出現
Abstract:
Deep mutational scanning experiments aid in the surveillance and forecasting of viral evolution by providing prospective measurements of mutational effects on viral traits, but epistatic shifts in the impacts of mutations can hinder viral forecasting when measurements were made in outdated strain backgrounds.
ディープ変異スキャン実験は、ウイルス特性に対する変異の影響の予測的測定を提供することで、ウイルスの進化の監視と予測に役立ちますが、測定が古い株の背景で行われた場合、変異の影響のエピスタシスシフトがウイルスの予測を妨げる可能性があります。
Here, we report measurements of the impact of all single amino acid mutations on ACE2-binding affinity and protein folding and expression in the SARS-CoV-2 Omicron BA.2.86 spike receptor-binding domain (RBD).
ここでは、SARS-CoV-2 Omicron BA.2.86スパイク受容体結合ドメイン(RBD)におけるACE2結合親和性、タンパク質の折り畳みと発現に対するすべての単一アミノ酸変異の影響の測定結果を報告します。
As with other SARS-CoV-2 variants, we find a plastic and evolvable basis for receptor binding, with many mutations at the ACE2 interface maintaining or even improving ACE2-binding affinity.
他の SARS-CoV-2 変異体と同様に、受容体結合の可塑性があり進化可能な基盤が見つかり、ACE2 インターフェースの多くの変異が ACE2 結合親和性を維持または向上させています。
Despite its large genetic divergence, mutational effects in BA.2.86 have not diverged greatly from those measured in its Omicron BA.2 ancestor.
遺伝的差異が大きいにもかかわらず、BA.2.86 の変異の影響は、その祖先である Omicron BA.2 で測定されたものと大きく異なっていません。
However, we do identify strong positive epistasis among subsequent mutations that have accrued in BA.2.86 descendants.
しかし、BA.2.86 の子孫に生じたその後の変異には、強い正のエピスタシスが認められます。
Specifically, the Q493E mutation that decreased ACE2-binding affinity in all previous SARS-CoV-2 backgrounds is reversed in sign to enhance human ACE2-binding affinity when coupled with L455S and F456L in the currently emerging KP.3 variant.
具体的には、これまでのすべての SARS-CoV-2 背景で ACE2 結合親和性を低下させた Q493E 変異は、現在出現している KP.3 変異体で L455S および F456L と組み合わせると、符号が反転してヒト ACE2 結合親和性を高めます。
Our results point to a modest degree of epistatic drift in mutational effects during recent SARS-CoV-2 evolution but highlight how these small epistatic shifts can have important consequences for the emergence of new SARS-CoV-2 variants.
私たちの結果は、最近の SARS-CoV-2 の進化中に変異効果に若干のエピスタシスのドリフトが生じたことを示していますが、これらの小さなエピスタシスの変化が新しい SARS-CoV-2 変異体の出現に重要な影響を及ぼす可能性があることを浮き彫りにしています。
INTRODUCTION
The evolution of SARS-CoV-2 is marked by the continuous emergence of viral variants.
SARS-CoV-2 の進化は、ウイルス変異体の継続的な出現によって特徴づけられます。
Mutations in these variants often concentrate within the spike protein and in particular its receptor-binding domain (RBD), where they enable escape from neutralizing antibody immunity while maintaining or improving ACE2 receptor binding and cellular entry.
これらの変異体の変異は、スパイクタンパク質、特にその受容体結合ドメイン (RBD) に集中することが多く、ACE2 受容体結合と細胞侵入を維持または改善しながら、中和抗体免疫からの脱出を可能にします。
Though advances in genomic sequencing capacity have enabled real-time surveillance of the origin and spread of these viral variants, it remains difficult to rapidly determine the functional consequences of the mutations they sample.
ゲノム配列解析能力の進歩により、これらのウイルス変異体の起源と拡散をリアルタイムで監視することが可能になりましたが、サンプルとなる変異の機能的影響を迅速に判断することは依然として困難です。
Deep mutational scanning has emerged as a powerful experimental method to aid in viral surveillance.
ディープ変異スキャンは、ウイルス監視を支援する強力な実験方法として登場しました。
These experiments comprehensively measure the impacts of all single amino acid changes in the SARS-CoV-2 spike or RBD on key phenotypes such as receptor-binding affinity, cellular entry, and antibody escape.
これらの実験は、SARS-CoV-2 スパイクまたは RBD におけるすべての単一アミノ酸の変化が、受容体結合親和性、細胞侵入、抗体回避などの主要な表現型に与える影響を包括的に測定します。
Because these measurements are made prospectively (i.e. prior to the evolution of a variant), they can be immediately consulted upon discovery of a novel viral variant to provide preliminary insights into the properties it might exhibit.
これらの測定は前向きに(つまり、変異体の進化前に)行われるため、新しいウイルス変異体が発見されたらすぐに参照して、その変異体が示す可能性のある特性に関する予備的な洞察を得ることができます。
Deep mutational scanning can therefore provide early insights about a novel variant while more intensive retrospective analyses of variant phenotype proceed.
したがって、ディープ変異スキャンは、変異表現型のより集中的な遡及的分析が進む一方で、新しい変異に関する早期の洞察を提供することができます。
More recently, analysis of large deep mutational scanning datasets also shows promise in the forecasting and prediction of future viral evolution.
最近では、大規模なディープ変異スキャンデータセットの分析は、将来のウイルスの進化の予測と予言にも有望であることが示されています。
However, the impacts of mutations on protein function are not constant over time due to epistasis, the phenomenon where the functional impact of one mutation is modulated by another.
ただし、エピスタシス、つまり1つの変異の機能的影響が別の変異によって調整される現象のため、変異がタンパク質機能に与える影響は時間の経過とともに一定ではありません。
We and others have described important epistatic interactions shaping SARS-CoV-2 evolution, such as the interaction between N501Y and Q498R that facilitated the original emergence of Omicron and interactions between substitutions at positions 455, 456, and/or 493 that have shaped subsequent Omicron evolution.
私たちと他の研究者は、オミクロンの最初の出現を促進した N501Y と Q498R の相互作用や、その後のオミクロンの進化を形作った位置 455、456、および/または 493 の置換間の相互作用など、SARS-CoV-2 の進化を形作る重要なエピスタシス相互作用について説明しました。
The utility of deep mutational scanning data in SARS-CoV-2 surveillance and forecasting therefore depends on updated measurements of mutational effects within each major variant that emerges, as the effects of mutations measured in an earlier variant may not accurately portray the impact that mutation would have in the viruses that circulate at present.
したがって、SARS-CoV-2 の監視と予測におけるディープ ミューテーション スキャン データの有用性は、出現する各主要変異体内のミューテーション効果の最新測定に依存します。以前の変異体で測定されたミューテーション効果は、現在流行しているウイルスにミューテーションが与える影響を正確に表していない可能性があるためです。
Since the original emergence of Omicron BA.1 in November, 2021, a string of derivative Omicron lineages (e.g., BA.2, BA.4/BA.5, XBB.1.5) have continued to evolve and displace prior strains in a more or less linear fashion.
2021 年 11 月に Omicron BA.1 が最初に出現して以来、一連の派生 Omicron 系統 (BA.2、BA.4/BA.5、XBB.1.5 など) が進化を続け、以前の株をほぼ直線的に置き換えてきました。
However, in August, 2023, a new “saltation” variant dubbed BA.2.86 gathered attention due to its large number of sequence substitutions relative to concurrently circulating strains and its rapid detection across multiple countries around the world suggesting early spread.
しかし、2023年8月、BA.2.86と呼ばれる新しい「跳躍」変異体が、同時に循環していた株と比較して配列置換の数が多く、世界中の複数の国で急速に検出されたことから早期拡散を示唆し、注目を集めました。
BA.2.86 and the predominant strains in August, 2023 such as XBB.1.5, EG.5 (XBB.1.5 + F456L) or HK.3 (EG.5 + L455F, i.e. “FLip”) last shared a common ancestor in BA.2, meaning BA.2.86 had been evolving for >1 year separately from the competing strains, allowing it to sample unique mutations such as a single amino acid deletion at position 483 (near major antibody epitopes) and the presence of a novel glycan at position 354.
BA.2.86と、2023年8月に優勢だったXBB.1.5、EG.5(XBB.1.5 + F456L)、HK.3(EG.5 + L455F、つまり「FLip」)などの株は、最後にBA.2で共通の祖先を共有しました。つまり、BA.2.86は競合する株とは別に1年以上進化しており、位置483(主要な抗体エピトープの近く)での単一のアミノ酸欠失や位置354での新規グリカンの存在など、独自の変異をサンプリングすることができました。
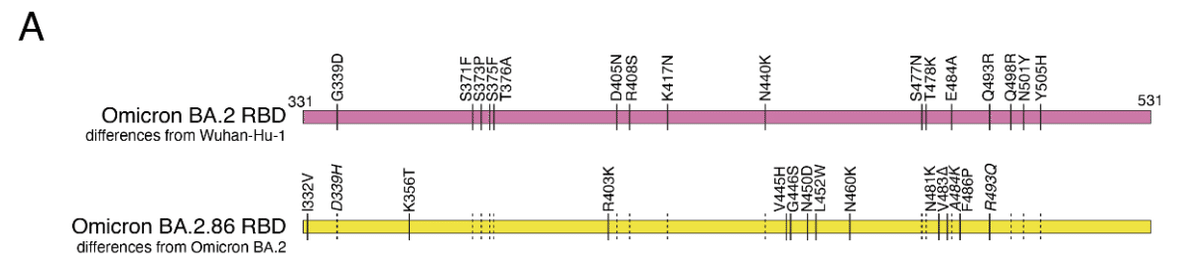
Due to the large number of sequence substitutions in BA.2.86 (34 spike mutations relative to BA.2, and 14 in the RBD, Figure 1A), there was immediate concern whether it would have a similar advantage in antibody-escape over competing XBB.1.5 variants as the original Omicron strain exhibited compared to pre-Omicron variants.
BA.2.86 には多数の配列置換 (BA.2 と比較して 34 個のスパイク変異、RBD では 14 個、図 1A) があるため、元の Omicron 株がプレ Omicron 変異体と比較して示したように、競合する XBB.1.5 変異体に対して抗体回避において同様の利点があるかどうかがすぐに懸念されました。
However, BA.2.86 was soon found to exhibit a similar degree of resistance to serum antibodies as the competing XBB.1.5 descendants.
しかし、BA.2.86 はすぐに、競合する XBB.1.5 子孫と同程度の血清抗体に対する耐性を示すことがわかりました。
Nonetheless, BA.2.86 did show a slight transmission advantage over co-circulating strains, and following acquisition of the L455S mutation in JN.1 that further optimized antibody evasion, rose to dominate case counts in December, 2023.
それにもかかわらず、BA.2.86 は共存する株よりもわずかに伝染力に優れており、抗体回避をさらに最適化する JN.1 の L455S 変異の獲得に続いて、2023 年 12 月には症例数で圧倒的な数にまで増加しました。
JN.1 is now spinning out further derivatives (e.g., KP.3) exhibiting enhanced fitness, suggesting future SARS-CoV-2 evolution will continue for some time to stem from the BA.2.86 evolutionary lineage.
JN.1 は現在、強化された適応度を示すさらなる派生株 (KP.3 など) を生み出しており、将来の SARS-CoV-2 の進化は BA.2.86 の進化系統に由来してしばらく続くことを示唆しています。
To aid in ongoing viral surveillance and forecasting, here we report deep mutational scanning data in the BA.2.86 RBD.
進行中のウイルス監視と予測を支援するために、ここでは BA.2.86 RBD の詳細な変異スキャン データを報告します。
We measure the impacts of all single amino acid changes and single-codon deletions in the BA.2.86 RBD on ACE2-binding affinity and RBD folding/expression, revealing continued tolerance to mutation within this viral domain.
BA.2.86 RBD のすべての単一アミノ酸の変化と単一コドンの欠失が ACE2 結合親和性と RBD の折り畳み/発現に与える影響を測定し、このウイルス ドメイン内での変異に対する耐性が継続していることを明らかにしました。
We find that mutational effects in BA.2.86 largely resemble those as measured in BA.2 despite their large sequence divergence.
BA.2.86 の変異の影響は、配列が大きく異なるにもかかわらず、BA.2 で測定されたものとほぼ類似していることがわかりました。
Nonetheless, we find a key epistatic interaction between Q493E and mutations at positions 455 and 456 that support the currently emerging KP.3 sub-variant.
それにもかかわらず、現在出現している KP.3 サブバリアントをサポートする位置 455 と 456 の変異と Q493E の間に重要なエピスタシス相互作用があることがわかりました。
RESULTS
Deep mutational scanning of the Omicron BA.2.86 RBD
Omicron BA.2.86 RBDの詳細な変異スキャン
We have previously described a yeast-surface display platform for deep mutational scanning of the SARS-CoV-2 RBD.
我々は以前、SARS-CoV-2 RBD の詳細な変異スキャンのための酵母表面ディスプレイ プラットフォームについて説明しました。
We have used this platform to measure the impacts of mutations in the Wuhan-Hu-1, Alpha, Beta, Delta, Eta, BA.1, BA.2, BQ.1.1, and XBB.1.5 strains on ACE2-binding affinity and RBD folding efficiency.
このプラットフォームを使用して、Wuhan-Hu-1、Alpha、Beta、Delta、Eta、BA.1、BA.2、BQ.1.1、XBB.1.5 株の変異が ACE2 結合親和性と RBD フォールディング効率に与える影響を測定しました。
Here, we follow this pipeline to generate mutational data in the SARS-CoV-2 BA.2.86 variant RBD background.
ここでは、このパイプラインに従って、SARS-CoV-2 BA.2.86 変異体 RBD バックグラウンドで変異データを生成します。
We first created a site-saturation mutagenesis library introducing every possible single amino acid mutation across the BA.2.86 RBD.
まず、BA.2.86 RBD 全体にわたって考えられるすべての単一アミノ酸変異を導入する部位飽和変異ライブラリを作成しました。
We also programmed single-codon deletions at each position, as well as premature stop codons or silent encoding of the wildtype codon at a fraction of sites as internal controls.
また、各位置で単一コドンの削除、および一部のサイトでの野生型コドンの未熟な終止コドンまたはサイレントエンコードを内部コントロールとしてプログラムしました。
We cloned library mutants together with an variant-identifier N16 nucleotide barcode, and we used PacBio long-read sequencing to link library barcodes with their associated RBD mutant and survey the composition of our library.
ライブラリ変異体を変異体識別子 N16 ヌクレオチド バーコードとともにクローン化し、PacBio ロングリード シーケンシングを使用してライブラリ バーコードを関連する RBD 変異体にリンクし、ライブラリの構成を調査しました。
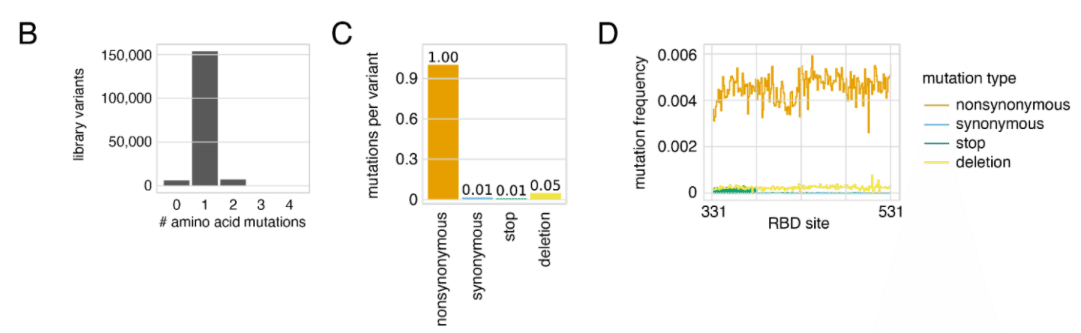
Our library showed the expected balance of single-mutant variants (Figure 1B) of the expected types (Figure 1C), with even mutation rates across the 200 RBD positions (Figure 1D).
ライブラリは、予想されるタイプ (図 1C) の単一変異体変異体 (図 1B) の予想通りのバランスを示し (図 1D)、200 個の RBD 位置にわたって変異率が均一でした (図 1D)。
We transformed the BA.2.86 RBD mutant library into a yeast-surface display platform that enables genotype-phenotype linkage between the mutant plasmid a yeast contains and the folded RBD protein that it expresses tethered to its cell surface.
BA.2.86 RBD 変異体ライブラリを酵母表面ディスプレイ プラットフォームに変換し、酵母に含まれる変異体プラスミドと、細胞表面に結合して発現する折り畳まれた RBD タンパク質との間の遺伝子型と表現型のリンクを可能にしました。
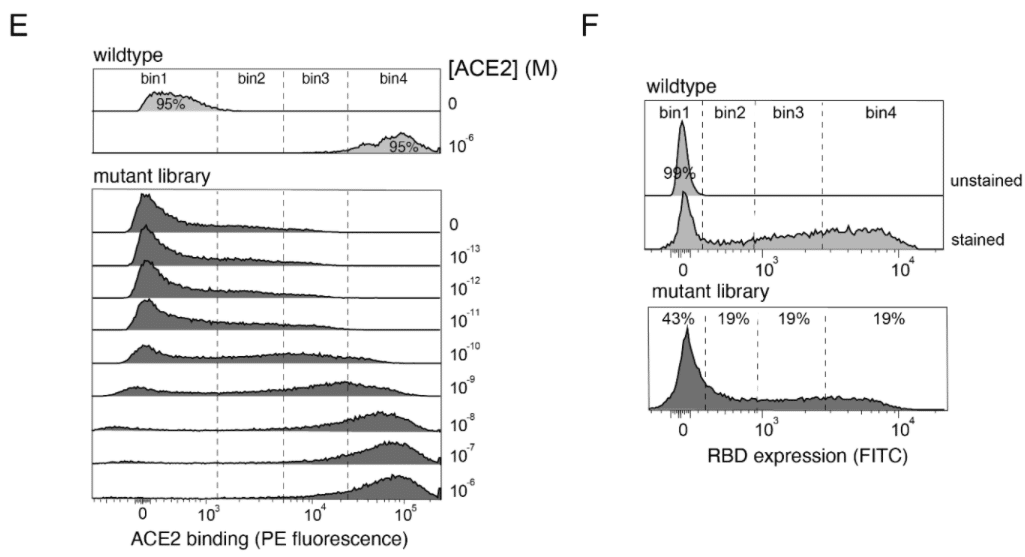
We incubated yeast-displayed RBD libraries across a concentration gradient of fluorescently-labeled monomeric human ACE2 or an antibody targeting a C-terminal c-Myc tag, and we used fluorescence-activated cell sorting (FACS) to partition library variants on the basis of activity (Figure 1E,F).
酵母ディスプレイ RBD ライブラリを、蛍光標識された単量体ヒト ACE2 または C 末端 c-Myc タグを標的とする抗体の濃度勾配で培養し、蛍光活性化細胞選別 (FACS) を使用して、活性に基づいてライブラリ変異体を分割しました (図 1E、F)。
We deep sequenced the linked barcodes in each FACS bin and determined the impact of each mutation on ACE2-binding affinity or RBD expression level based on the distribution of sequencing counts across FACS bins.
各 FACS ビンのリンクされたバーコードをディープシーケンスし、FACS ビン全体のシーケンスカウントの分布に基づいて、各変異が ACE2 結合親和性または RBD 発現レベルに与える影響を判定しました。
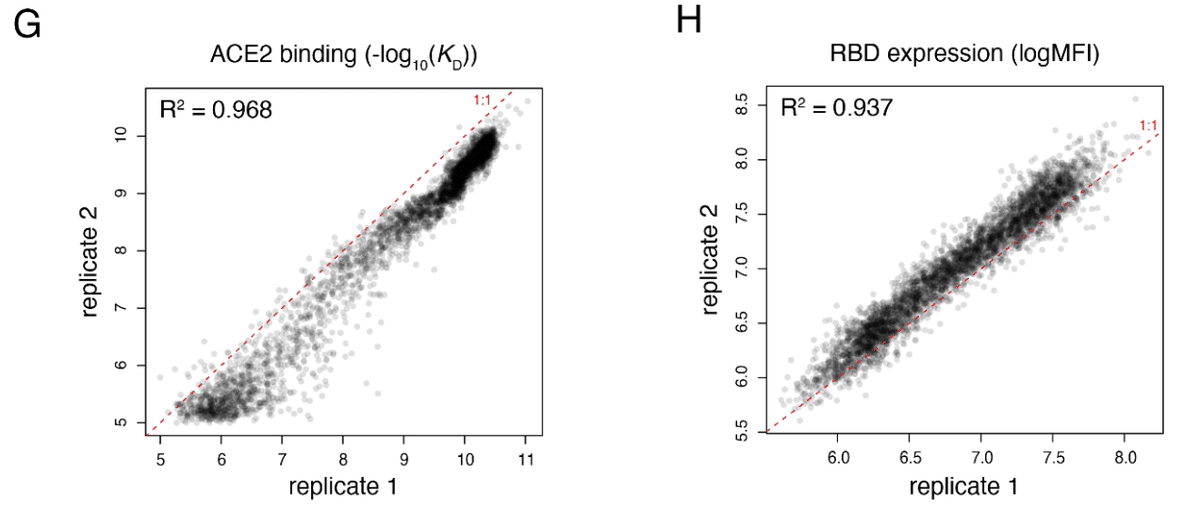
These deep mutational scanning measurements were conducted in duplicate with independently barcoded variant libraries, with per-mutant phenotypes strongly correlated between replicates (Figures 1G, H).
これらのディープ変異スキャン測定は、独立してバーコード化された変異ライブラリを使用して 2 回実施され、変異体ごとの表現型は複製間で強く相関していました (図 1G、H)。
Heatmaps illustrating the impact of each RBD mutation on ACE2-binding affinity and RBD expression are presented in Figure 2.
図 2 に、各 RBD 変異が ACE2 結合親和性と RBD 発現に与える影響を示すヒートマップを示します。
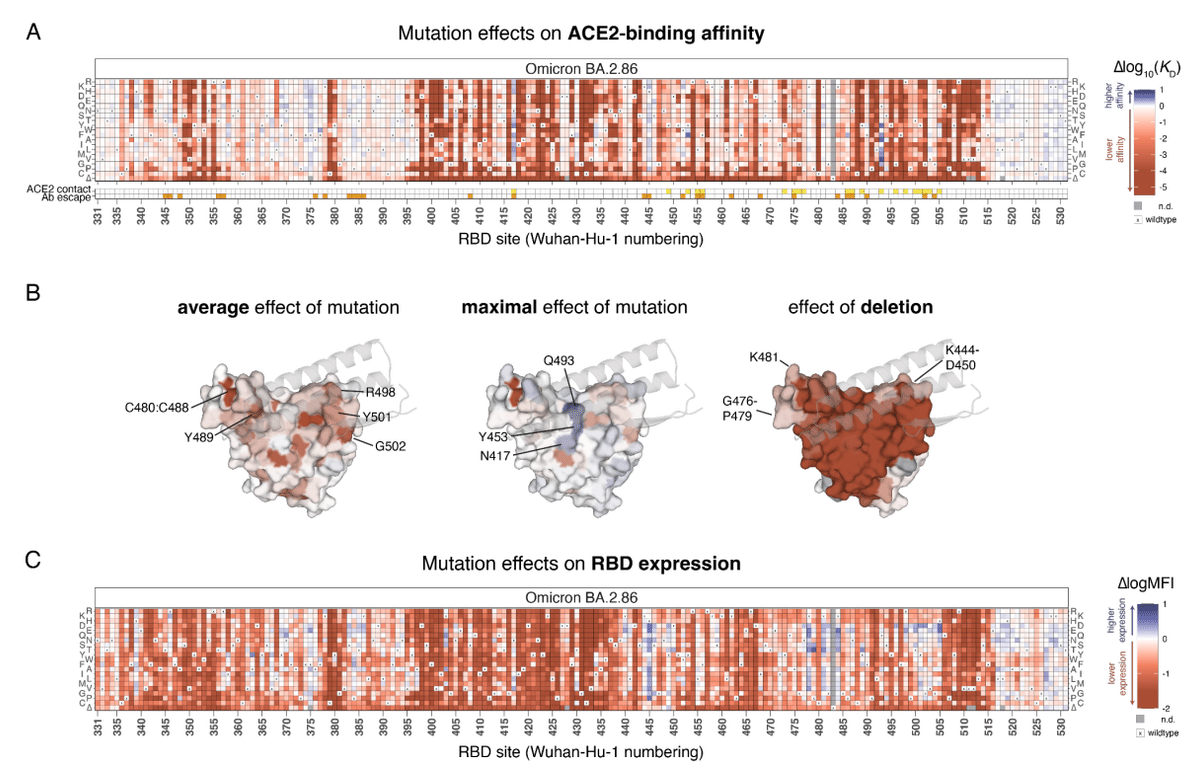
To aid visualization of these large datasets, interactive heatmaps, including measurements from prior SARS-CoV-2 variants, are available at https://tstarrlab.github.io/SARS-CoV-2-RBD_DMS_Omicron-EG5-FLip-BA286/RBD-heatmaps/.
これらの大規模なデータセットの視覚化を支援するために、以前の SARS-CoV-2 変異体の測定値を含むインタラクティブなヒートマップが https://tstarrlab.github.io/SARS-CoV-2-RBD_DMS_Omicron-EG5-FLip-BA286/RBD-heatmaps/ で入手できます。
As with deep mutational scans of prior SARS-CoV-2 variants, we find considerable tolerance to mutation in the RBD, with many amino acid mutations having neutral or positive impacts on ACE2-binding affinity, including those that directly contact ACE2 (indicated by yellow squares, Figure 2A).
以前の SARS-CoV-2 変異体の詳細な変異スキャンと同様に、RBD の変異に対する耐性はかなり高く、ACE2 に直接接触するもの (黄色の四角で表示、図 2A) を含め、多くのアミノ酸変異が ACE2 結合親和性に中立またはプラスの影響を与えています。
Although some direct contact residues are highly constrained (e.g., Y489 [Wuhan-Hu-1 reference numbering used throughout], R498, Y501, and G502; Figure 2B, left), many contact residues are tolerant to mutation and can sample affinity-enhancing mutations (e.g., N417, Y453, Q493; Figure 2B, center).
直接接触する残基の中には、非常に制約されているものもありますが (例: Y489 [Wuhan-Hu-1 参照番号は全体を通して使用]、R498、Y501、G502、図 2B 左)、多くの接触残基は変異に対して耐性があり、親和性を高める変異をサンプリングできます (例: N417、Y453、Q493、図 2B 中央)。
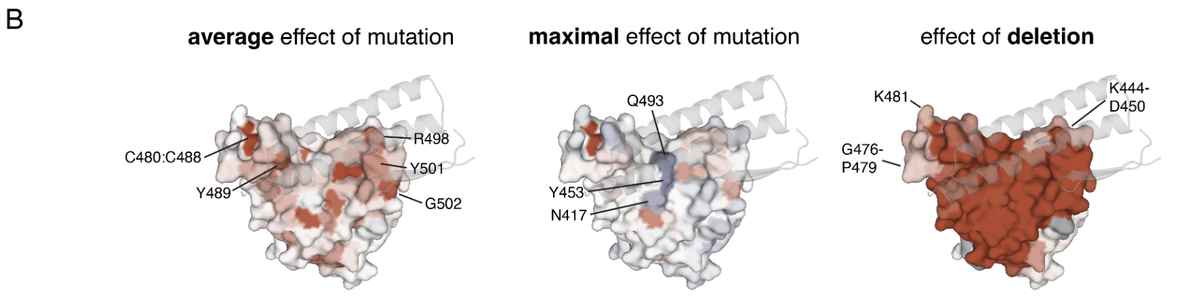
Deletions at the ACE2 interface are primarily deleterious, though two peripheral loops where indels occur more broadly during sarbecovirus evolution are less constrained (Figure 2B, right).
ACE2 インターフェースでの欠失は主に有害ですが、サルベコウイルスの進化中にインデルがより広範囲に発生する 2 つの周辺ループはそれほど制約されていません (図 2B、右)。
(Taylor & Starr 2023). Our experiments also identify mutations that would enhance RBD expression (Figure 2C), a proxy for folding and stability.
私たちの実験では、折り畳みと安定性の指標となる RBD 発現 (図 2C) を強化する変異も特定されています。
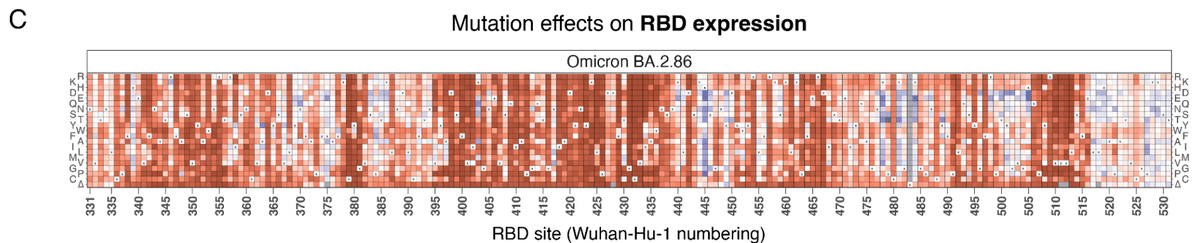
Many of these stabilizing mutations remove basic residues at the ACE2 interface that have been introduced in variant evolution since Wuhan-Hu-1, for example mutations to H445 (originally V445 in Wuhan-Hu-1), K478 (originally T478), K481 (originally N481), and K484 (originally E484).
これらの安定化変異の多くは、武漢-Hu-1 以降の変異進化で導入された ACE2 インターフェースの塩基性残基を除去します。たとえば、H445 (武漢-Hu-1 では元々 V445)、K478 (元々 T478)、K481 (元々 N481)、K484 (元々 E484) への変異です。
The increase in positively charged residues at the ACE2 interface conferred by these basic BA.2.86 residues has been noted to increase electrostatic complementarity with the corresponding ACE2 surface that is rich in acidic residues, which our data suggest may come at some cost for isolated RBD folding efficiency.
これらの塩基性 BA.2.86 残基によってもたらされる ACE2 インターフェースの正電荷残基の増加は、酸性残基が豊富な対応する ACE2 表面との静電的相補性を高めることが知られています。これは、私たちのデータから、孤立した RBD 折り畳みの効率にいくらかの犠牲を払う可能性があることが示唆されています。
We previously identified stabilizing space-filling mutations that compensate for a loss of lipid tail volume that occupies space in the RBD core in the locked spike trimer structure; our deep mutational scanning map in BA.2.86 shows similar patterns of space-filling stabilizing mutations in the RBD core (e.g., I358F, A363Y), suggesting that the positive impact that these mutations had on RBD vaccine stability in earlier variants will continue in vaccine designs derived from BA.2.86.
我々は以前、ロックされたスパイク三量体構造の RBD コアの空間を占める脂質尾部の容積の減少を補う安定化空間充填変異を特定しました;BA.2.86 の深部変異スキャンマップは、RBD コアの空間充填安定化変異の同様のパターン (I358F、A363Y など) を示しており、これらの変異が以前の変異体の RBD ワクチンの安定性に与えたプラスの影響が、BA.2.86 から派生したワクチン設計でも継続することを示唆しています。
Epistatic shifts in mutational effects
突然変異の影響におけるエピスタシスの変化
We next determined the extent to which the effects of mutations in the BA.2.86 RBD differ compared to their effects in the BA.2 ancestor due to epistasis.
次に、BA.2.86 RBD の変異の影響が、エピスタシスにより BA.2 祖先における影響と比較してどの程度異なるかを調べました。
We computed a sitewise “epistatic shift” metric for each RBD residue, which identifies sites where the effects of mutations on ACE2-binding affinity differ more or less strongly between a pair of variants (Figure 3; interactive visualization of all variants and sites available at https://tstarrlab.github.io/SARS-CoV-2-RBD_DMS_Omicron-EG5-FLip-BA286/epistatic-shifts/).
各 RBD 残基について、部位ごとの「エピスタシス シフト」メトリックを計算しました。これは、変異の ACE2 結合親和性への影響が、変異のペア間で多かれ少なかれ大きく異なる部位を特定します (図 3、すべての変異と部位のインタラクティブな視覚化は、https://tstarrlab.github.io/SARS-CoV-2-RBD_DMS_Omicron-EG5-FLip-BA286/epistatic-shifts/ で利用できます)。
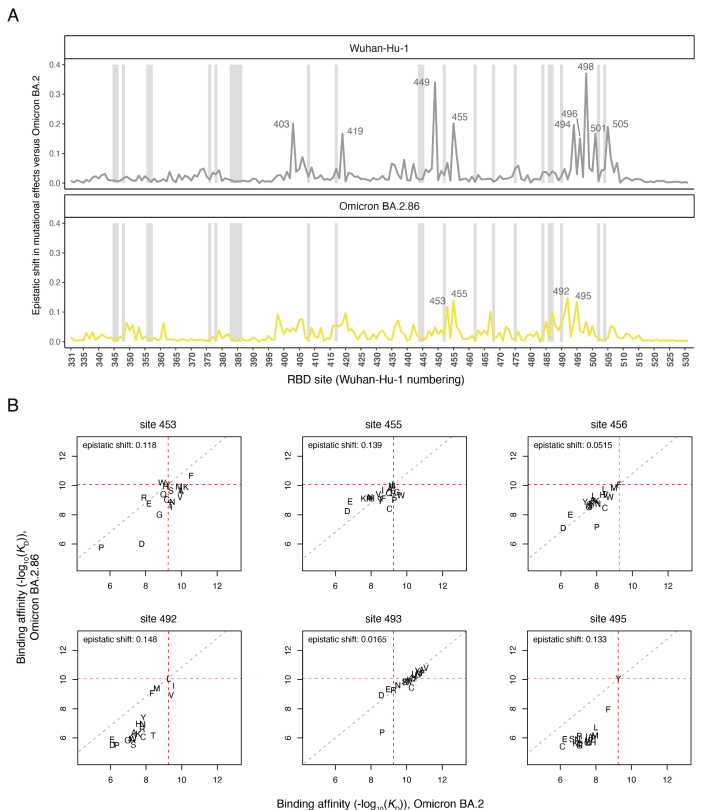
The epistatic shift is a probabilistic distance metric (Jensen-Shannon distance) that compares sitewise profiles of mutational effects between two variant backgrounds, computed on the vectors of affinities measured for the 20 amino acids possible at a position.
エピスタシス シフトは、2 つの変異体背景間の変異効果のサイトごとのプロファイルを比較する確率的距離メトリック (Jensen-Shannon 距離) であり、位置で可能な 20 個のアミノ酸について測定された親和性のベクトルに基づいて計算されます。
The epistatic shift metrics scales from 0 to 1, with 0 indicating a site where the measured affinities of each amino acid mutant are identical between backgrounds and 1 indicating a site where the distributions are entirely dissimilar.
エピスタシス シフト メトリックは 0 から 1 の範囲で、0 は各アミノ酸変異体の測定された親和性が背景間で同一であるサイトを示し、1 は分布が完全に異なるサイトを示します。
We previously found that the 16 substitutions separating Omicron BA.2 from the ancestral Wuhan-Hu-1 variant induced strong and widespread epistatic shifts across the RBD’s ACE2-contact interface (Figure 3A, top), largely attributed to the impact of the N501Y substitution.
我々は以前、Omicron BA.2 と祖先の Wuhan-Hu-1 変異体を隔てる 16 個の置換が、RBD の ACE2 接触界面全体で強力かつ広範囲のエピスタシス シフトを引き起こした (図 3A、上部) ことを発見しました。これは主に N501Y 置換の影響によるものです。
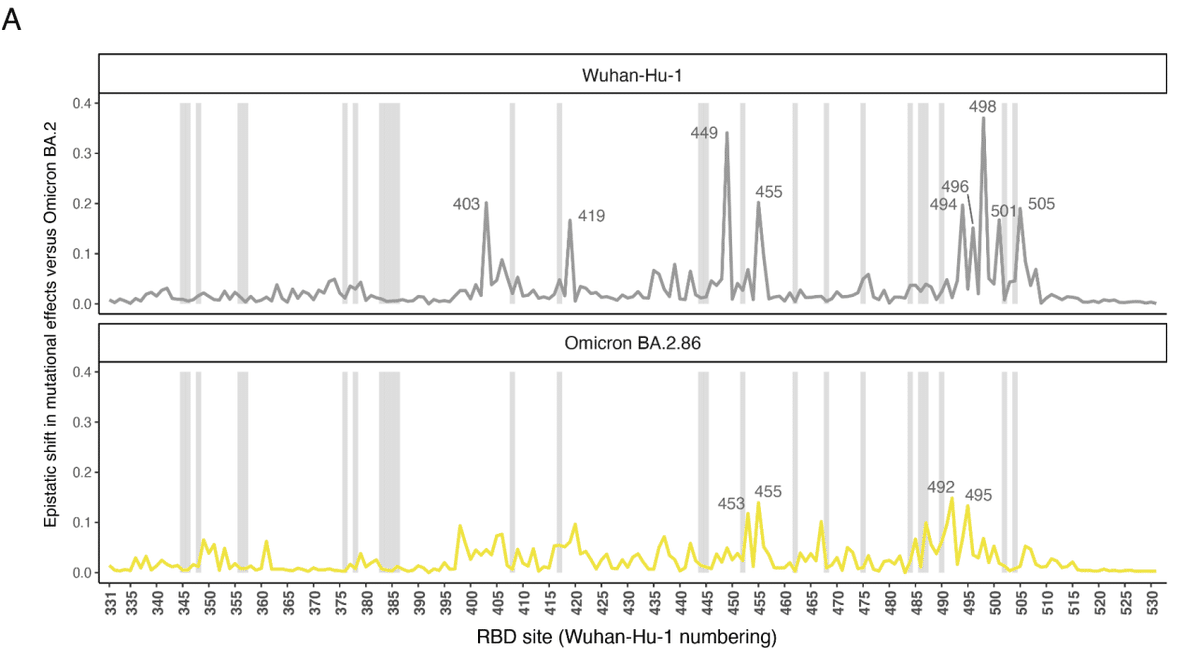
BA.2.86 is separated from BA.2 by a similar number of RBD changes (14 changes) as BA.2 itself is separated from Wuhan-Hu-1 (Figure 1A), raising the question whether similarly dramatic epistatic shifts are present between BA.2 and BA.2.86.
BA.2.86 は、BA.2 自体が Wuhan-Hu-1 から分離されているのと同数の RBD 変化 (14 の変化) によって BA.2 から分離されています (図 1A)。このことから、BA.2 と BA.2.86 の間にも同様に劇的なエピスタシスの変化が存在するかどうかという疑問が生じます。
The sitewise epistatic shifts between BA.2.86 and BA.2, however, are notably modest compared to the epistatic shifts seen between Wuhan-Hu-1 and BA.2 (Figure 3A, bottom).
ただし、BA.2.86 と BA.2 の間のサイトごとのエピスタシスの変化は、Wuhan-Hu-1 と BA.2 の間に見られるエピスタシスの変化と比較すると、著しく控えめです (図 3A、下)。
Among the sites with the strongest epistatic shifts (Figure 3B), the more common trend is that mutations that enhanced ACE2 affinity in the BA.2 background become constrained in BA.2.86 (lower-right quadrant of scatterplots in Figure 3B, e.g. V453, W455, or I492) instead of mutations that were deleterious in BA.2 becoming affinity-enhancing in BA.2.86 (upper-right quadrant, e.g. W453).
最も強いエピスタシスシフトを示す部位(図 3B)では、BA.2 バックグラウンドで ACE2 親和性を高めた変異が BA.2.86 で制約を受ける傾向(図 3B の散布図の右下象限、例: V453、W455、または I492)が一般的であり、BA.2 で有害であった変異が BA.2.86 で親和性を高める傾向(右上象限、例: W453)は見られません。
Despite showing few newly expanded pathways for ACE2-affinity enhancement, BA.2.86 itself has been found to have tighter binding affinity for ACE2 compared to BA.2 and competing XBB.1.5 subvariants, which also expands pathways of evolution by offsetting mutations like L455S that trade off improved antibody escape with slightly reduced ACE2-binding affinity.
ACE2 親和性を高めるための新たに拡張された経路はほとんど示されていないにもかかわらず、BA.2.86 自体は、BA.2 や競合する XBB.1.5 サブバリアントと比較して、ACE2 に対する結合親和性が高いことがわかっており、これにより、抗体の回避の改善と ACE2 結合親和性のわずかな低下をトレードオフする L455S などの変異を相殺することで、進化の経路も拡張されます。
Epistatic emergence of the KP.3 variant
KP.3変異体のエピスタシス的出現
Following the initial discovery of BA.2.86, its rise in global frequency was relatively slow until derivative lineages acquired additional spike changes that increased fitness.
BA.2.86 が最初に発見された後、派生系統が適応度を高める追加のスパイク変化を獲得するまで、その世界的な頻度の上昇は比較的緩やかでした。
The first BA.2.86 descendant to rise to global dominance and displace the competing XBB.1.5-related variants was JN.1 (BA.2.86 + L455S), and as of July, 2024, KP.3 variants which also sample F456L and Q493E (Figure 4A) comprise the majority of global sequences.
世界的な優位性を獲得し、競合する XBB.1.5 関連変異体を置き換えた最初の BA.2.86 子孫は JN.1 (BA.2.86 + L455S) であり、2024 年 7 月現在、F456L と Q493E もサンプリングする KP.3 変異体 (図 4A) が世界的な配列の大部分を占めています。
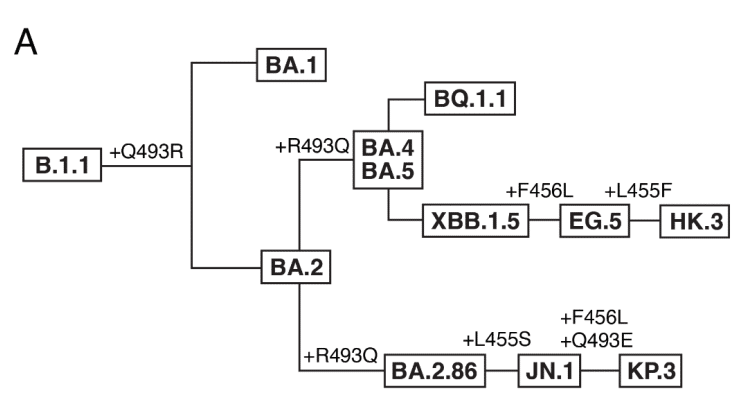
The Q493E mutation in KP.3 presented as a particular surprise, as this mutation has been strongly deleterious to ACE2 binding throughout SARS-CoV-2 variant evolution, decreasing affinity by approximately one order of magnitude in previously measured backgrounds and decreasing affinity ~5.4-fold in the BA.2.86 data reported here.
KP.3 の Q493E 変異は特に驚きでした。この変異は SARS-CoV-2 変異体の進化を通じて ACE2 結合に非常に有害であり、以前に測定されたバックグラウンドでは親和性が約 1 桁低下し、ここで報告された BA.2.86 データでは親和性が約 5.4 倍低下したからです。
We have previously described strong epistatic interactions between position 493 with positions 455 and 456, and so we wondered whether the deleterious effect of Q493E was modulated by the co-occurring L455S and/or F456L mutations.
我々は以前、位置 493 と位置 455 および 456 の間に強いエピスタシス相互作用があることを説明したため、Q493E の有害な影響が、同時に発生する L455S および/または F456L 変異によって調整されるのではないかと考えました。
To address this question, we measured ACE2 binding in isogenic yeast-display titration assays for the L455S, F456L, and Q493E mutations in single, double, and triple mutant combinations (Figure 4B).
この疑問に対処するために、単一、二重、および三重変異体の組み合わせにおける L455S、F456L、および Q493E 変異について、同質酵母ディスプレイ滴定アッセイで ACE2 結合を測定しました (図 4B)。
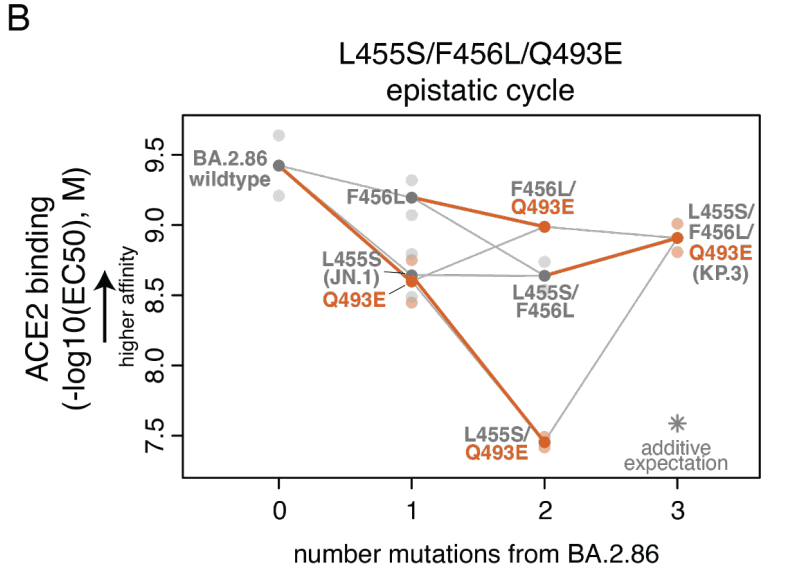
We found that the Q493E mutation is affinity-decreasing on all backgrounds tested except when sampled together with both L455S and F456L, where the effect of Q493E switches in sign to be affinity-enhancing.
Q493E 変異は、L455S と F456L の両方と一緒にサンプリングされた場合を除いて、テストしたすべての背景で親和性を低下させることがわかりました。L455S と F456L の両方と一緒にサンプリングされた場合、Q493E の効果は親和性を高める方向に反転します。
This result is consistent with recent surface plasmon resonance data.
この結果は、最近の表面プラズモン共鳴データと一致しています。
This strong positive epistasis between L455S, F456L, and Q493E allows KP.3 to maintain ACE2 binding affinity between the levels of JN.1 and BA.2.86, which coupled with its increased immune evasiveness compared to JN.1, likely explains its current rise in frequency.
L455S、F456L、Q493E 間のこの強い正のエピスタシスにより、KP.3 は JN.1 と BA.2.86 のレベル間で ACE2 結合親和性を維持できます。これは、JN.1 と比較して免疫回避性が高まっていることと相まって、現在の頻度の上昇を説明していると考えられます。
This second observation of strong epistasis between positions 455, 456, and 493 is intriguing in light of the large sequence variation at these positions that occurs over broader sarbecovirus evolution (Figure 4C).
位置 455、456、493 間のこの 2 回目の強いエピスタシスの観察は、より広範なサルベコウイルスの進化を通じてこれらの位置で発生する大きな配列変異を考慮すると興味深いものです (図 4C)。
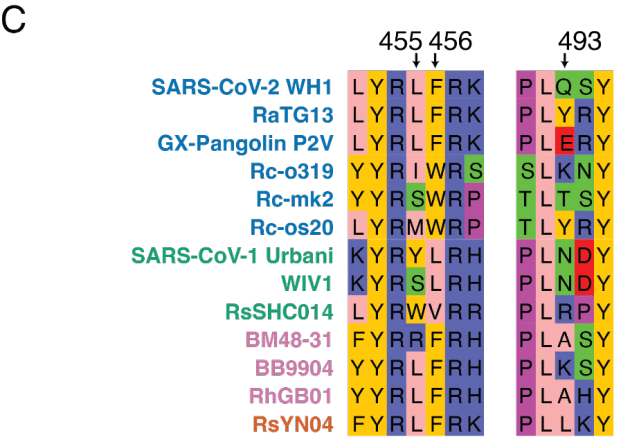
Of note, the GX-Pangolin virus, which can bind strongly and enter efficiently via human ACE2, also naturally samples the Q493E mutation despite sharing the L455 and F456 residues with ancestral SARS-CoV-2 variants like Wuhan-Hu-1 where Q493E is highly detrimental.
注目すべきは、ヒトACE2を介して強力に結合し、効率的に侵入できるGX-Pangolinウイルスは、L455およびF456残基を、Q493Eが非常に有害であるWuhan-Hu-1などの祖先SARS-CoV-2変異体と共有しているにもかかわらず、Q493E変異も自然にサンプリングするということです。
The presence of Q493E in the GX-Pangolin RBD may therefore point to alternative epistatic interactions that can also compensate for Q493E outside of changes to residues 455 or 456, highlighting the plastic nature of mutational effects in sarbecovirus RBDs.
したがって、GX-Pangolin RBDにQ493Eが存在することは、残基455または456の変化以外でもQ493Eを補うことができる代替のエピスタシス相互作用を示している可能性があり、サルベコウイルスRBDにおける変異効果の可塑性を浮き彫りにしています。
DISCUSSION
Here we report deep mutational scanning measurements of the impact of mutations in the BA.2.86 RBD on ACE2 binding and folded RBD expression.
ここでは、BA.2.86 RBD の変異が ACE2 結合と折り畳まれた RBD 発現に与える影響について、詳細な変異スキャン測定を報告します。
We anticipate ongoing utility of these mutational maps in evolutionary forecasting and surveillance of SARS-CoV-2 evolution.
これらの変異マップは、SARS-CoV-2 の進化の進化予測と監視において今後も有用であると期待しています。
Our measurements reveal only modest epistatic shifts in mutational effects between BA.2 and BA.2.86 despite a similar number of substitutions separating these variants as separates BA.2 from the ancestral Wuhan-Hu-1 RBD.
我々の測定では、BA.2 と BA.2.86 の間では、BA.2 と祖先の Wuhan-Hu-1 RBD を分けるのと同数の置換がこれらの変異体を分けているにもかかわらず、変異効果のエピスタシスシフトはわずかであることが明らかになった。
The strong and widespread epistatic shifts caused by N501Y during early SARS-CoV-2 variant evolution therefore continues to be an anomaly in its scale.
したがって、SARS-CoV-2 変異体の初期の進化中に N501Y によって引き起こされた強力で広範なエピスタシスシフトは、その規模において依然として異常である。
Nonetheless, small-scale epistatic shifts have also proven important in variant emergence, where unexpected mutations like Q493E (which contributes to KP.3’s enhanced antibody escape) remain capable of occurring on variant backgrounds that epistatically optimize their functional impacts.
それにもかかわらず、小規模なエピスタシスの変化も変異体の出現において重要であることが証明されており、Q493E(KP.3 の抗体回避の強化に寄与)のような予期しない変異は、機能的影響をエピスタシス的に最適化する変異体背景で発生する可能性があります。
We have now described two different sets of sites where the impacts of mutations are highly context dependent.
私たちは、変異の影響が状況に大きく依存する 2 つの異なる部位のセットについて説明しました。
The first, demonstrated here and in prior publication, comprises residues 455, 456, and 493 in the central beta-strand of the RBD ACE2-contact surface.
1 つ目は、ここで示され、以前の論文でも示されているように、RBD ACE2 接触面の中央のベータ ストランドにある残基 455、456、493 で構成されています。
The other, demonstrated by us and others in prior work, comprises residues 498 and 501.
もう 1 つは、以前の研究で私たちと他の人が示したように、残基 498 と 501 で構成されています。
All five of these residues are hotspots of evolution in SARS-CoV-2 as well as the broader sarbecovirus lineage, but the strong epistasis among these positions likely limits our ability to predict what new combinations are compatible with human ACE2 binding.
これら 5 つの残基はすべて、SARS-CoV-2 だけでなく、より広範なサルベコウイルス系統の進化のホットスポットですが、これらの位置間の強いエピスタシスにより、どのような新しい組み合わせがヒト ACE2 結合と互換性があるかを予測する能力が制限される可能性があります。
Future work to exhaustively characterize epistatic interaction among the entire suite of amino acids that can occur at these positions would therefore have great utility for ongoing evolutionary modeling of SARS-CoV-2 and sarbecovirus evolution.
したがって、これらの位置で発生する可能性のあるアミノ酸群全体のエピスタシス相互作用を徹底的に特徴付ける今後の研究は、SARS-CoV-2 とサルベコウイルスの進化の進行中の進化モデル化に非常に役立つでしょう。
以下省略。
この記事が気に入ったらサポートをしてみませんか?