XBB 系統のスパイク構造、立体構造、抗原性、および受容体認識

SARS-CoV-2 Omicron XBB lineage spike structures, conformations, antigenicity, and receptor recognition
SARS-CoV-2 Omicron XBB 系統のスパイク構造、立体構造、抗原性、および受容体認識
Summary
A recombinant lineage of the SARS-CoV-2 Omicron variant, named XBB, appeared in late 2022 and evolved descendants that successively swept local and global populations.
XBB と名付けられた SARS-CoV-2 オミクロン変異体の組換え系統は 2022 年後半に出現し、子孫を進化させ、地域および世界の人口を次々と席巻しました。
XBB lineage members were noted for their improved immune evasion and transmissibility.
XBB 系統のメンバーは、免疫回避性と伝染性が向上していることで注目されました。
Here, we determine cryo-EM structures of XBB.1.5, XBB.1.16, EG.5 and EG.5.1 spike (S) ectodomains to reveal reinforced 3-RBD-down receptor inaccessible closed states mediated by interprotomer receptor binding domain (RBD) interactions previously observed in BA.1 and BA.2.
ここでは、XBB.1.5、XBB.1.16、EG.5、および EG.5.1 スパイク (S) 外部ドメインの低温 EM 構造を決定し、強化された 3-RBD ダウン受容体がアクセスできない閉鎖状態を明らかにします。これは、BA.1 および BA.2 で以前に観察されたプロトマー間受容体結合ドメイン (RBD) 相互作用によって媒介されます。
Improved XBB.1.5 and XBB.1.16 RBD stability compensated for stability loss caused by early Omicron mutations, while the F456L substitution reduced EG.5 RBD stability.
XBB.1.5 および XBB.1.16 RBD の安定性の向上により、初期の Omicron 変異による安定性の損失が補われましたが、F456L 置換により EG.5 RBD の安定性が低下しました。
Improved stability of XBB.1.5 and XBB.1.16 RBD compensated for loss of stability caused by early Omicron mutations, while the F456L substitution reduced EG.5 RBD stability.
XBB.1.5 および XBB.1.16 RBD の安定性の向上により、初期の Omicron 変異による安定性の損失が補われました。 一方、F456L 置換は EG.5 RBD の安定性を低下させました。
S1 subunit mutations had long-range impacts on conformation and epitope presentation in the S2 subunit.
S1 サブユニットの変異は、S2 サブユニットの立体構造とエピトープ提示に広範囲の影響を及ぼしました。
Our results reveal continued S protein evolution via simultaneous optimization of multiple parameters including stability, receptor binding and immune evasion, and the dramatic effects of relatively few residue substitutions in altering the S protein conformational landscape.
我々の結果は、安定性、受容体結合、免疫回避を含む複数のパラメータの同時最適化によるSタンパク質の継続的な進化と、Sタンパク質の立体構造の変化における比較的少数の残基置換の劇的な効果を明らかにしました。
Introduction
Since its discovery in November 2021, the SARS-CoV-2 Omicron B.1.1.529 variant, noted for its high number of mutations in the spike (S) protein relative to previous variants of concern (VOC), has continued to mutate and has diversified into many sublineages that have gone through a series of global shifts in dominance (Figure S1).
SARS-CoV-2 オミクロン B.1.1.529 変異株は、2021 年 11 月に発見されて以来、以前の懸念される変異株 (VOC) と比較して、スパイク (S) タンパク質の変異数が多いことで注目されてきました。 変異を続け、一連の世界的な優位性の変化を経て多くの亜系統に多様化しました(図S1)。
Identified in late 2022, the XBB lineage, a recombinant of two highly diversified BA.2 sublineages, became the first documented example of a SARS-CoV-2 variant increasing its fitness through recombination rather than substitutions.
2022 年後半に同定された、高度に多様化した 2 つの BA.2 亜系統の組換え体である XBB 系統は、SARS-CoV-2 変異株が置換ではなく組換えによって適応度を高めた最初の文書化された例となった。
XBB.1, the early XBB lineage member that gave rise to the initial expansion of the XBB lineages, was remarkably resistant to antiviral humoral immunity induced by vaccination or breakthrough infections of prior Omicron subvariants, with its immune resistant properties attributed to multiple receptor binding domain (RBD) substitutions and the Y144del N-terminal domain (NTD) mutation that has been recurrently observed and originally noted in the Alpha VOC.
XBB.1 は、XBB 系統の初期拡大を引き起こした初期の XBB 系統メンバーであり、ワクチン接種または以前の Omicron 亜変異体のブレイクスルー感染によって誘導される抗ウイルス液性免疫に対して顕著な耐性を示しました。 その免疫抵抗特性は、複数の受容体結合ドメイン (RBD) 置換と、繰り返し観察され、もともとアルファ VOC で注目されていた Y144del N 末端ドメイン (NTD) 変異に起因すると考えられます。
XBB.1 was more infectious than BA.2 in pseudovirus infectivity assays and its RBD bound to ACE2 stronger than BA.2 RBD.
XBB.1 はシュードウイルス感染力アッセイにおいて BA.2 よりも感染力が高く、その RBD は BA.2 RBD よりも強く ACE2 に結合しました。
While significantly more fusogenic than BA.2 and BA.2.75, XBB.1 pathogenicity was comparable to BA.2.75 and less than Delta.
BA.2 および BA.2.75 よりも著しく融合性が高い一方で、XBB.1 の病原性は BA.2.75 に匹敵し、デルタ株よりも低かった。
XBB.1.5, a descendent of XBB.1 harboring an additional F486P substitution in the RBD, was identified around October 2022.
RBD に追加の F486P 置換を含む XBB.1 の子孫である XBB.1.5 は、2022 年 10 月頃に特定されました。
XBB.1.5 became the most prevalent lineage worldwide outcompeting the previously most prevalent Omicron BA.5 descendent BQ.1.1 variant (Figure S1).
XBB.1.5 は、以前は最も普及していた Omicron BA.5 の子孫である BQ.1.1 亜種を上回り、世界中で最も普及した系統になりました (図 S1)。
As XBB.1.5 began to decline by mid-summer 2023, XBB.1.16 together with a complex array of additional minor lineages replaced it.
XBB.1.5 は 2023 年半ばまでに衰退し始めたため、XBB.1.16 と追加のマイナー リネージの複雑な配列がそれに置き換わりました。
Around July 2023, EG.5 began to rapidly spread in some Asian and North American countries.
2023 年 7 月頃、EG.5 はアジアと北米の一部の国で急速に普及し始めました。
EG.5’s higher effective reproduction number (R0) than XBB.1.9.2 (parental with same S protein amino acid sequence as XBB.1.5), XBB.1.5 and XBB.1.16, attributed to its enhanced immune evasion properties, contributed to its global spread.
EG.5 の実効再生産数 (R0) は、XBB.1.9.2 (XBB.1.5 と同じ S タンパク質アミノ酸配列を持つ親)、XBB.1.5、および XBB.1.16 よりも高く、免疫回避特性が強化されたため、世界的な蔓延に貢献したと考えられています。
EG.5.1, the immediate descendent of EG.5, had one additional Q52H substitution in the NTD, and was the EG.5 sublineage that spread most rapidly.
EG.5 の直接の子孫である EG.5.1 は、NTD 内にさらに 1 つの Q52H 置換を持ち、最も急速に拡散した EG.5 亜系統でした。
As expected from their sequence similarities, the S proteins of XBB lineage members, XBB.1.5, XBB.1.16 and EG.5, cluster together on a phylogenetic tree and are distinct from other VOCs and from their parental BA.2 (Figure S2).
配列類似性から予想されるように、XBB 系統メンバー、XBB.1.5、XBB.1.16、および EG.5 の S タンパク質は、 系統樹上にクラスターを形成し、他の VOC やその親 BA.2 とは区別されます (図 S2)。
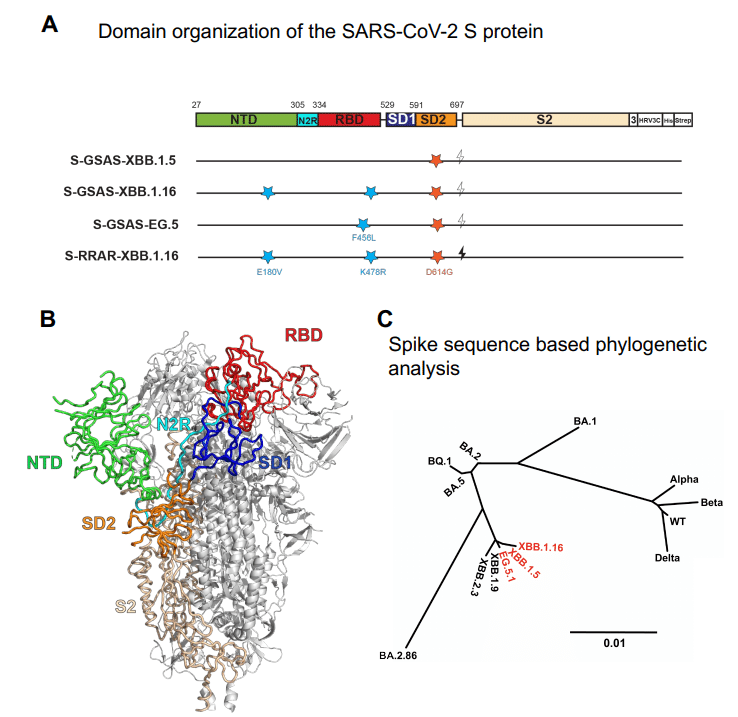
Given the lead time required for vaccine evaluation and manufacture, XBB.1.5, the dominant variant in the spring of 2023, was selected as the S variant to be used as a monovalent booster for the vaccine update (Pfizer, Moderna, Novavax) that become available in the fall of 2023.
ワクチンの評価と製造に必要なリードタイムを考慮すると、 2023 年春の主要なバリアントである XBB.1.5 は、2023 年の秋に利用可能になるワクチン更新プログラム (ファイザー、モデルナ、ノババックス) の一価ブースターとして使用される S バリアントとして選択されました。
Early studies have shown the booster to be protective against currently circulating variants.
初期の研究では、ブースターが現在流行している変異種に対して保護効果があることが示されています。
The S protein on the SARS-CoV-2 surface, composed of S1 and S2 subunits, is the primary determinant of its receptor binding and immune evasion properties (Figure S2).
S1 サブユニットと S2 サブユニットで構成される SARS-CoV-2 表面の S タンパク質は、その受容体結合と免疫回避特性の主な決定因子です (図 S2)。
The S1 subunit is responsible for receptor binding and forms a cap over the fusion subunit S2 in the pre-fusion conformation of the S protein.
S1 サブユニットは受容体結合を担当し、S タンパク質の融合前立体構造において融合サブユニット S2 の上にキャップを形成します。
The S1 NTD subunit (residues 27–305) is connected via a linker (N2R; residues 306–334) to the RBD (residues 335–521), followed by the subdomain 1 (SD1; residues 529–591) and subdomain 2 (SD2; residues 592–697) regions.
S1 NTD サブユニット (残基 27 ~ 305) はリンカー (N2R; 残基 306 ~ 334) を介して RBD (残基 335 ~ 521) に接続されています。続いてサブドメイン 1 (SD1; 残基 529 ~ 591) 領域とサブドメイン 2 (SD2; 残基 592 ~ 697) 領域が続きます。
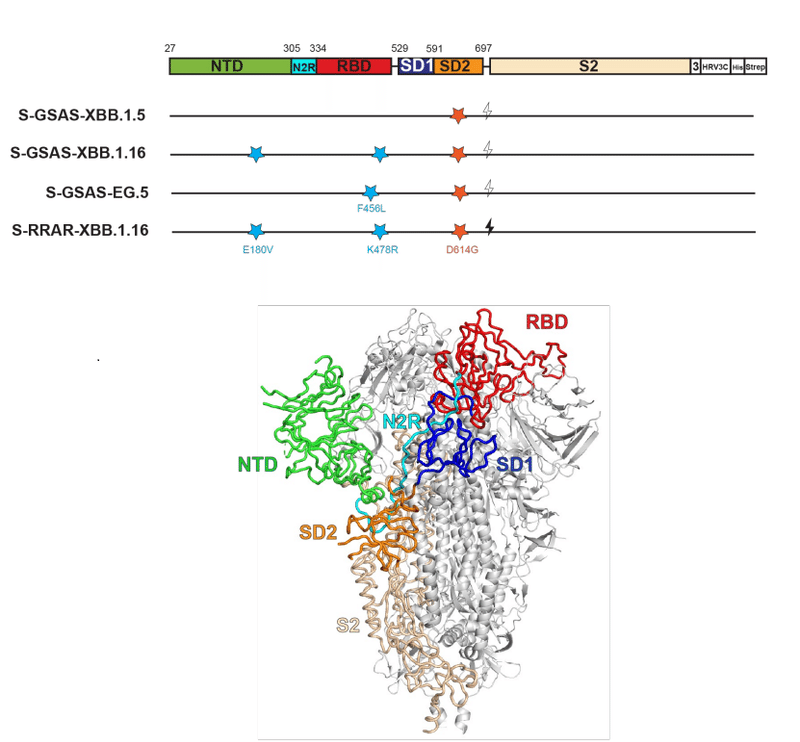
The RBD contains a receptor binding motif (RBM; residues 483–506) that binds host receptors including ACE2 and TMEM106B.
RBD には、ACE2 や TMEM106B などの宿主受容体に結合する受容体結合モチーフ (RBM; 残基 483 ~ 506) が含まれています。
The RBD adopts either a receptor-inaccessible “down” or closed conformation, or a receptor-accessible “up” or open conformation.
RBDは、受容体がアクセスできない「ダウン」または閉じた立体構造、または受容体がアクセスできる「アップ」または開いた立体構造のいずれかを採用します。
SD2 harbors a furin clevage site that defines the boundary between the S1 and S2 subunits.
SD2 には、S1 サブユニットと S2 サブユニットの間の境界を定義するフューリン切断部位が存在します。
Another cleavage site S2’ is located in the S2 subunit.
別の切断部位 S2’ は S2 サブユニットにあります。
Following receptor binding and proteolytic processing of the S protein, the S2 subunit reorganizes upon detachment of S1, allowing for insertion of the hydrophobic fusion peptide into the host membrane and transition of the S2 helices to the postfusion conformation, leading to cell entry.
受容体結合と S タンパク質のタンパク質分解プロセシングに続いて、S2 サブユニットは S1 の分離時に再構成され、疎水性融合ペプチドの宿主膜への挿入と S2 ヘリックスの融合後立体構造への移行を可能にし、細胞侵入につながります。
The early Omicron variants BA.1 and BA.2 acumulated stabilizing mutations at the RBD-RBD interprotomer contacts in the receptor-inaccessible down state, resulting in a shift of the S protein population towards greater occupancy of the receptor-inaccesible 3-RBD-down state, either through direct interactions between the RBDs or by stabilizing structural features involved in interprotomer RBD interactions.
初期の Omicron 変異体 BA.1 および BA.2 は、受容体がアクセスできないダウン状態で RBD-RBD インタープロトマー接触部に安定化変異を蓄積しました。 その結果、RBD間の直接相互作用またはプロトマー間RBD相互作用に関与する構造的特徴の安定化によって、受容体がアクセスできない3-RBDダウン状態をより多く占める方向へSタンパク質集団がシフトします。
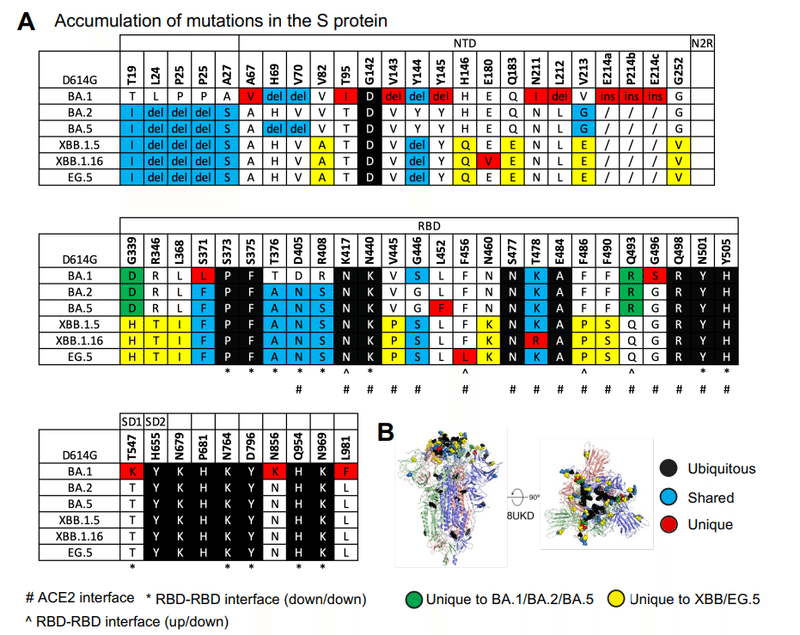
These mutations are retained in the XBB.1.5, XBB.1.16 and EG.5 S proteins (Figures 1A and S2).
これらの変異は、XBB.1.5、XBB.1.16、および EG.5 S タンパク質に保持されています (図 1A および S2)。
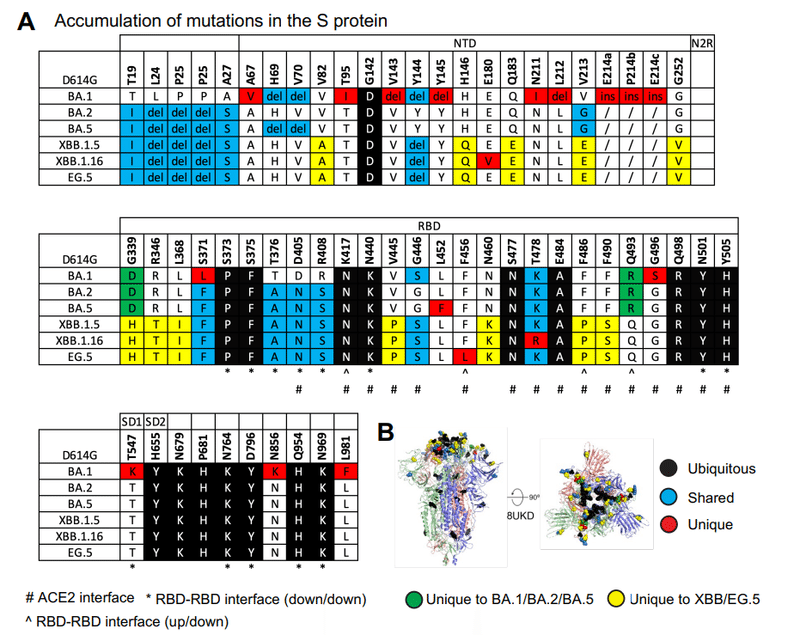
To understand how the S protein structures changed in these evolved Omicron subvariants, here we determine cryo-EM structures and study the receptor binding, antigenicity and stability of XBB.1.5, and of the closely related XBB.1.16 and EG.5 and EG.5.1 S ectodomains using our previously established S-GSAS-D614G (uncleaved) or S-RRAR-D614G (furin-cleaved) platforms prepared without the incorporation of any proline stabilization mutation, as well as a construct expressing only the RBD of each variant (Figure S2, Data S1 and S2).
これらの進化した Omicron サブバリアントで S タンパク質の構造がどのように変化したかを理解するために、ここでは、クライオ EM 構造を決定し、XBB.1.5 の受容体結合、抗原性、安定性を研究します。プロリン安定化変異を組み込むことなく調製された、以前に確立された S-GSAS-D614G (非切断) または S-RRAR-D614G (フリン切断) プラットフォームを使用した、密接に関連する XBB.1.16 および EG.5及びEG.5.1 S 外部ドメイン、 各変異体の RBD のみを発現する構築物も同様に研究します(図 S2、データ S1 および S2)。
Results
Results Thermostability of the XBB.1.5, XBB.1.16 and EG5 RBD
XBB.1.5、XBB.1.16、および EG5 RBD の熱安定性
New mutations in the XBB.1.5, XBB.1.16, EG.5 and EG.5.1 S proteins relative to the original Omicron lineages BA.1, BA.2 and BA.5 are located in the NTD and RBD, with several appearing at the ACE2 interacting site (Figures 1A and B).
元の Omicron 系統 BA.1、BA.2、および BA.5 と比較した XBB.1.5、XBB.1.16、EG.5、および EG.5.1 S タンパク質の新しい変異は、NTD および RBD に位置します。いくつかはACE2相互作用部位に現れます(図1AおよびB)。
We have previously reported dramatic loss in BA.1 RBD stability relative to the WT RBD, with subsequent BA.2 RBD mutations partially recovering RBD stability.
我々は以前、WT RBDと比較してBA.1 RBDの安定性が劇的に低下し、その後のBA.2 RBD変異によりRBDの安定性が部分的に回復することを報告した。
To assess the impact of the additional acquired RBD mutations on the stability of the domain, we used a differential scanning fluorimetry (DSF) assay that measures changes in protein intrinsic fluorescence as a thermal ramp is applied to the sample (Figure 1C and S3).
追加で獲得した RBD 変異がドメインの安定性に及ぼす影響を評価するために、サンプルに熱ランプを適用した際のタンパク質固有蛍光の変化を測定する示差走査蛍光分析 (DSF) アッセイを使用しました (図 1C および S3)。
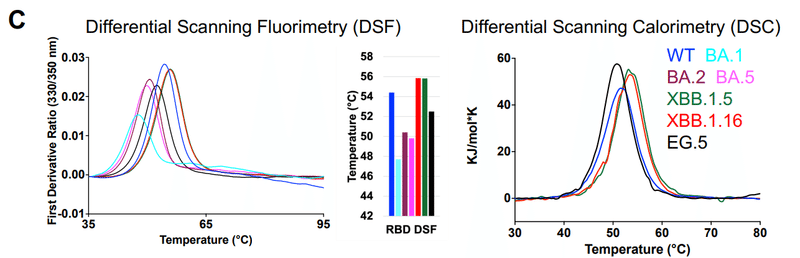
The BA.1 RBD showed an inflection temperature (Ti) of 47.6 ± 0.2 °C, ~7 °C lower than that of the WT RBD, similar to previous reports.
BA.1 RBD は、以前のレポートと同様に、47.6 ± 0.2 °C の変曲点 (Ti) を示し、WT RBD よりも約 7 °C 低くなりました。
This loss in thermostability was partially recovered in the BA.2 RBD (Ti of 50.6 ± 0.1 °C).
この熱安定性の損失は、BA.2 RBD (Ti 50.6 ± 0.1 °C) では部分的に回復しました。
XBB.1.5 showed higher thermostability than the WT RBD with a Ti of ~55.8 °C.
XBB.1.5 は、Ti が ~55.8 °C の WT RBD よりも高い熱安定性を示しました。
Thus, mutations acquired in XBB.1.5 not only recovered the reduced thermostability observed in the early Omicron subvariants, but the XBB.1.5 RBD also surpassed the thermostability of the WT RBD.
したがって、XBB.1.5 で獲得された突然変異は、初期のオミクロン株で観察された熱安定性の低下を回復しただけでなく、XBB.1.5 RBD は WT RBD の熱安定性も上回りました。
The RBDs of XBB.1.5 and XBB.1.16 differ by a single residue substitution at position 478.
XBB.1.5 と XBB.1.16 の RBD は、478 位の単一残基置換が異なります。
A T478K substitution that occurred in Omicron BA.1 was retained in BA.2, BA.5 and XBB.1.5, but was substituted to an Arginine in XBB.1.16 (Figure 1A and S2).
Omicron BA.1 で発生した T478K 置換は、BA.2、BA.5、および XBB.1.5 では保持されましたが、XBB.1.16 ではアルギニンに置換されました (図 1A および S2)。
As the DSF profiles of XBB.1.5 and XBB.1.16 RBDs were nearly identical (Figure 1C), we concluded that the K478R substitution, which is the only differing residue between the XBB.1.5 and XBB.1.16 RBDs, did not impact RBD thermostability.
XBB.1.5 と XBB.1.16 RBD の DSF プロファイルはほぼ同一であったため (図 1C)、われわれは、XBB.1.5 と XBB.1.16 RBD の間で唯一異なる残基である K478R 置換は RBD の熱安定性に影響を及ぼさないと結論付けました。
The EG.5 RBD showed a Ti of 52.5 ± 0.1 °C, which was ~1.8 °C lower than that of the WT RBD and ~3 °C lower than XBB.1.5 and XBB.1.16 RBD, implicating F456L, the only residue substitution between XBB.1.5 and EG.5, in the reduced stability of the EG.5 RBD.
EG.5 RBD は、52.5 ± 0.1 °C の Ti を示しました。これは、WT RBD の Ti よりも約 1.8 °C 低く、XBB.1.5 および XBB.1.16 RBD よりも約 3 °C 低かったです。 これは、XBB.1.5 と EG.5 の間の唯一の残基置換である F456L が、EG.5 RBD の安定性の低下に関与していることを示しています。
Melting temperatures (Tm) of WT, XBB.1.5, XBB.1.6 and EG.5 RBD measured using Differential Scanning Calorimetry (DSC), showed the same thermostability trends as observed in the DSF experiments, i.e., EG.5 < WT < XBB.1.5 ~XBB.1.16 (Figure 1C and S3A).
示差走査熱量測定 (DSC) を使用して測定された WT、XBB.1.5、XBB.1.6 および EG.5 RBD の融解温度 (Tm) は、DSF 実験で観察されたのと同じ熱安定性傾向、つまり EG.5 < WT < XBB.1.5 ~ XBB.1.16 を示しました (図 1C および S3A)。
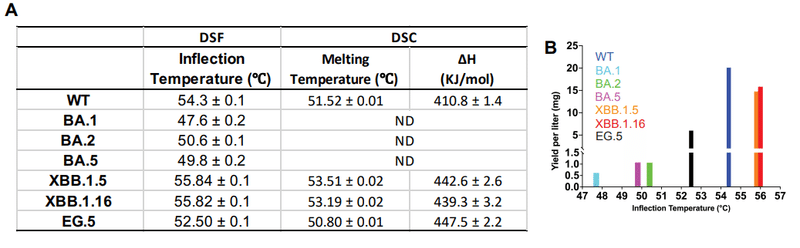
The highest protein yields were obtained from the XBB.1.5 and XBB.1.16 RBD constructs, with both also being the most thermostable of the panel tested, while the BA.1 RBD was both the least thermostable and also gave the lowest in expression yield (Figure S3B).
最高のタンパク質収量は XBB.1.5 および XBB.1.16 RBD コンストラクトから得られ、どちらもテストしたパネルの中で最も熱安定性が高くなります。 一方、BA.1 RBD は熱安定性が最も低く、発現収率も最も低かった (図 S3B)。
In summary, RBD thermostability varied during SARS-CoV-2 Omicron evolution.
要約すると、RBD の熱安定性は SARS-CoV-2 オミクロンの進化中に変化しました。
A drop in thermostability observed with the Omicron BA.1 RBD, was progressively recovered in subsequent variants.
Omicron BA.1 RBD で観察された熱安定性の低下は、後続のバリエーションでは徐々に回復しました。
XBB.1.5 and XBB.1.16 RBDs were more thermostable than the WT RBD, while EG.5 showed a reduction in thermostabilty compared to WT RBD, although it still remained more thermostable than BA.1, BA.2 and BA.5 RBDs.
XBB.1.5 および XBB.1.16 RBD は WT RBD よりも熱安定性が高かったのに対し、EG.5 は WT RBD と比較して熱安定性の低下を示しましたが、依然として BA.1、BA.2、および BA.5 RBD よりも熱安定性が高いままでした。
Stabilization of the RBD may lead to its more productive encounter with host receptor thereby improving virus transmissibility.
RBD の安定化は、RBD と宿主受容体とのより生産的な出会いをもたらし、それによってウイルスの伝播性を改善する可能性があります。
Since the RBD is one of the most immunodominant regions of the S protein, its stability may also have vaccine implications, with improved stability resulting in a more durable immunogen, both in the S protein and RBD-only vaccine formats.
RBD は S タンパク質の最も免疫優勢な領域の 1 つであるため、その安定性はワクチンにも影響を与える可能性があり、S タンパク質と RBD のみのワクチン形式の両方において、安定性の向上によりより耐久性の高い免疫原が得られます。
ACE2 receptor binding of XBB lineage S protein ectodomains
XBB系統Sタンパク質細胞外ドメインのACE2受容体結合
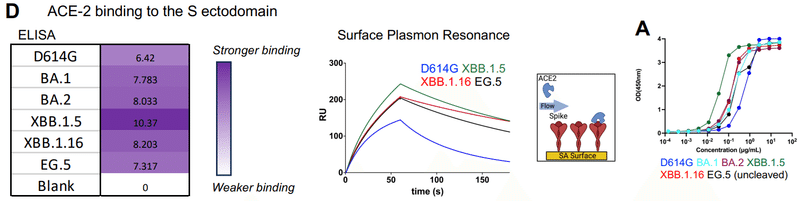
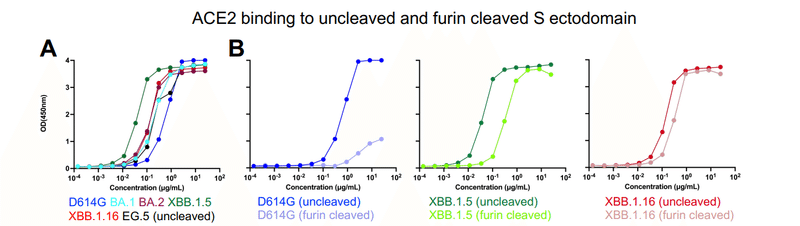
To assess the impact of the acquired mutations on receptor engagement, we measured binding of the S protein ectodomains to the ACE2 receptor ectodomain by ELISA and Surface Plasmon Resonance (SPR) (Figures 1D, 2A-G and S3C).
受容体結合に対する獲得変異の影響を評価するために、ELISAおよび表面プラズモン共鳴(SPR)によって、ACE2受容体外部ドメインへのSタンパク質外部ドメインの結合を測定しました(図1D、2A〜GおよびS3C)。
As expected based on previous studies, despite extensive mutation of the ACE2 binding region of the RBD, all the S proteins retained robust binding to ACE2.
以前の研究に基づいて予想されたように、RBD の ACE2 結合領域の広範な変異にもかかわらず、すべての S タンパク質は ACE2 への強力な結合を保持していました。
Indeed, the binding levels for all the Omicron S proteins tested were higher than for the D614G S protein, with XBB.1.5 showing the strongest ACE2 binding (Figures 1D and 2A).
実際、試験したすべてのオミクロン S タンパク質の結合レベルは D614GS S タンパク質よりも高く、XBB.1.5 が最も強い ACE2 結合を示しました (図 1D および 2A)。
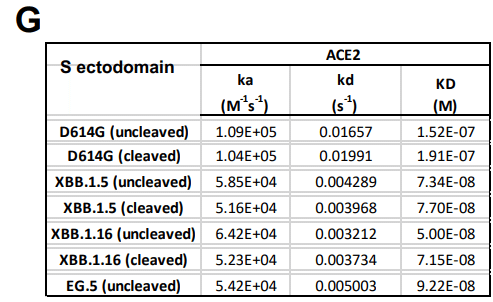
With the S protein tethered to the sensor surface via its Twin-Strep tag, using SPR, we measured ACE2 binding affinities (or dissociation constant; KD) of 152 nM and 192 nM for the uncleaved (S-GSAS) and furin-cleaved (S-RRAR) D614G spikes, respectively (Figures 2F-G).
SPR を使用して、Twin-Strep タグを介してセンサー表面に S タンパク質を結合すると、 非切断 (S-GSAS) およびフリン切断 (S-RRAR) D614G スパイクについて、それぞれ 152 nM および 192 nM の ACE2 結合親和性 (または解離定数; KD) を測定しました (図 2FG)。
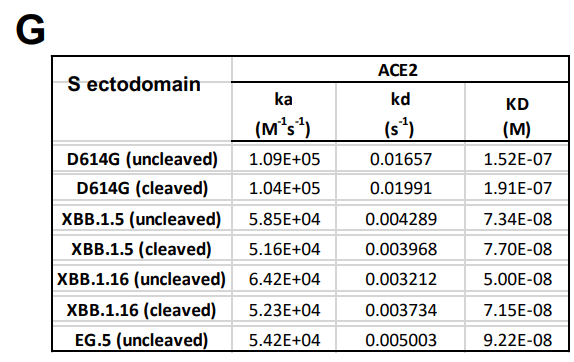
ACE2 affinities for the uncleaved S ectodomains of XBB.1.5, XBB.1.16 and EG.5 were 73 nM, 50 nM, and 92 nM, respectively, while ACE2 affinity of furin-cleaved S ectodomain of XBB.1.5 and XBB.1.16 were 74 nM and 71 nM, respectively.
XBB.1.5、XBB.1.16、および EG.5 の未切断の細胞外ドメインに対する ACE2 親和性は、それぞれ 73 nM、50 nM、および 92 nM でした。 一方、XBB.1.5 および XBB.1.16 のフューリン切断 S 外部ドメインの ACE2 親和性は、それぞれ 74 nM および 71 nM でした。
EG.5 and EG.5.1 S ectodomains bound ACE2 with similar affinity and kinetics (Figure S3E).
EG.5 および EG.5.1 S 細胞外ドメインは、同様の親和性および反応速度で ACE2 に結合しました (図 S3E)。
The tighter ACE2 affinity of the XBB.1.5, XBB.1.16, EG.5 and EG.5.1 S ectodomains compared to D614G was a result of the slower dissociation rates (koff) that also compensated for reduction in association rates (kon) (Figures 1D, 2C and 2F-G).
D614G と比較して、XBB.1.5、XBB.1.16、EG.5および EG.5.1 S 細胞外ドメインの ACE2 親和性がより強固であるのは、解離速度 (koff) が遅いためであり、会合速度 (kon) の低下も補われました。 (図 1D、2C、および 2F ~ G)。
ACE2 binding affinity and kinetics were similar for the uncleaved and furin-cleaved versions of the D614G, XBB.1.5 and XBB.1.16 S ectodomains.
ACE2 結合親和性と反応速度は、D614G、XBB.1.5、および XBB.1.16 S 外部ドメインの未切断バージョンとフリン切断バージョンで類似していました。
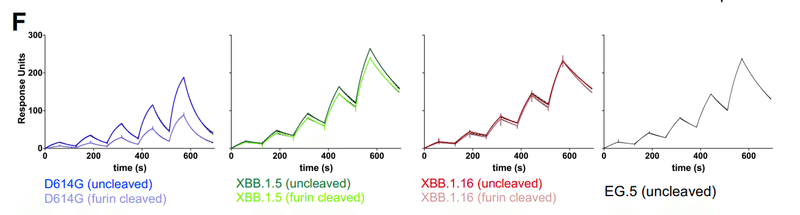
For the D614G variant, despite similar ACE2 binding affinities and kinetics, substantial and reproducible ACE2 binding level differences were observed between the furin-cleaved and uncleaved S proteins.
D614G バリアントでは、同様の ACE2 結合親和性と反応速度にも関わらず、フューリン切断 S タンパク質と非切断 S タンパク質の間で実質的かつ再現性のある ACE2 結合レベルの差が観察されました。
The uncleaved D614G S protein showed higher levels of ACE2 binding compared to the furin-cleaved D614G S protein, suggesting reduced availability of ACE2-accessible “up” RBD in the furin-cleaved S protein compared to the uncleaved version.
未切断の D614GS S タンパク質は、フリンで切断された D614GS S タンパク質と比較して、より高いレベルの ACE2 結合を示しました。これは、未切断のバージョンと比較して、フューリンで切断された S タンパク質では ACE2 がアクセス可能な「アップ」RBD の利用可能性が低下していることを示唆しています。
This observation is consistent with our previous reports of reduced RBD up populations in furin-cleaved D614G S compared to the uncleaved S ectodomain.
この観察は、未切断のS外部ドメインと比較して、フリン切断D614G S におけるRBDアップ集団の減少に関する我々の以前の報告と一致する。
By contrast, much less difference in binding levels was measured by SPR for the furin-cleaved versus uncleaved versions of the XBB.1.5 and XBB.1.16 S proteins, suggesting that the RBD up/down disposition in the XBB.1.5 and XBB.1.16 S proteins are less sensitive to furin-cleavage compared to the D614G S protein.
対照的に、XBB.1.5 および XBB.1.16 S タンパク質のフリン切断バージョンと非切断バージョンでは、SPR によって結合レベルの差ははるかに小さく測定されました。これは、XBB.1.5 および XBB.1.16 S タンパク質における RBD のup/downの性質が、D614GS S タンパク質と比較してフリン切断に対する感受性が低いことを示唆しています。
These differences between the uncleaved and furin-cleaved S ectodomains were reflected also in the ACE2 binding levels measured by ELISA (Figures 2A, B and E).
切断されていないS外部ドメインとフューリン切断されたS外部ドメインとの間のこれらの差異は、ELISAによって測定されたACE2結合レベルにも反映されていた(図2A、BおよびE)。
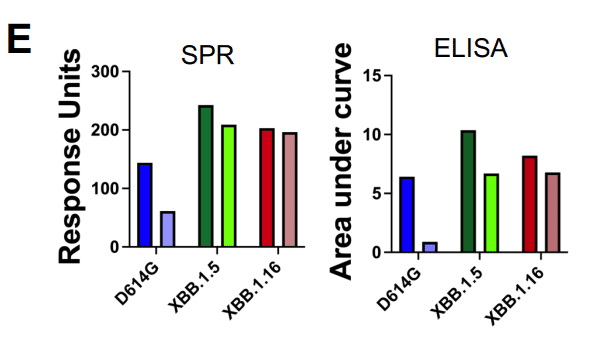
In summary, and consistent with published reports, our data demonstrated higher affinity binding of the ACE2 ectodomain to SARS-CoV-2 S ectodomains of XBB.1.5, XBB.1.16, EG.5 and EG.5.1 variants than D614G, contributed by reduced dissociation rates.
要約すると、公開されたレポートと一致して、我々のデータは、XBB.1.5、XBB.1.16、EG.5およびEG.5.1バリアントのSARS-CoV-2 S外部ドメインに対するACE2外部ドメインの結合親和性がD614Gよりも高いことを実証し、これは解離速度の低下に寄与した。
Further, differences in ACE2 binding levels observed for furin-cleaved versus uncleaved S ectodomains suggested a reduced impact of furin cleavage on domain dispositions of the XBB.1.5 and XBB.1.16 spikes than the D614G spike.
さらに、フューリン切断対非切断 S 外部ドメインで観察された ACE2 結合レベルの差は、XBB.1.5 および XBB.1.16 スパイクのドメイン性質に対するフューリン切断の影響が D614G スパイクよりも小さいことを示唆しました。
ACE2 receptor binding of XBB lineage RBDs
XBB系統RBDのACE2受容体結合
Within the context of the S protein, the RBD is subject to conformational masking and can adopt an “up” or receptor accessible state where the RBM is exposed and available for binding to the host receptor, or a receptor inaccessible “down” state where the RBM is conformationally masked.
S タンパク質の文脈内では、RBD は構造マスキングの影響を受けます。 そして、RBMが露出されて宿主受容体に結合できる「アップ」または受容体アクセス可能な状態、またはRBMが構造的にマスクされている受容体アクセス不可能な「ダウン」状態をとることができます。
To assess intrinsic binding of ACE2 to RBD without these conformational contraints, we measured ACE2 binding to a panel of RBD constructs derived from the D614G (or WT), BA.1, BA.2, BA.5, XBB.1.5, XBB.1.16 and EG.5/EG.5.1 S proteins (Figures 2H-I and S3C).
これらの立体構造上の制約なしにRBDへのACE2の固有の結合を評価するために、D614G (または WT)、BA.1、BA.2、BA.5、XBB.1.5、XBB.1.16、および EG.5/EG.5.1 S タンパク質に由来する RBD コンストラクトのパネルに対する ACE2 の結合を測定しました (図2H〜IおよびS3C)。
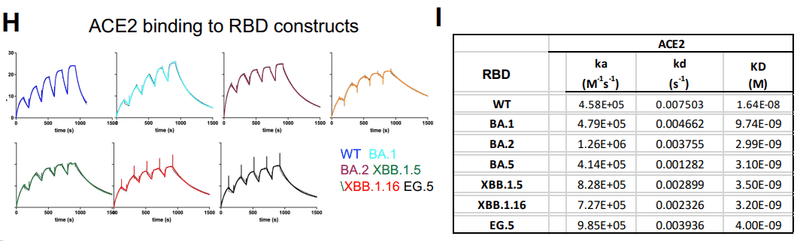
The WT and XBB.1.5 RBD bound ACE2 with affinities of ~16.4 nM and 3.5 nM, respectively, which are in close agreement with published studies.
WT および XBB.1.5 RBD は、それぞれ ~16.4 nM および 3.5 nM の親和性で ACE2 に結合しました。これは、公開された研究とほぼ一致しています。
The ACE2 affinity for the Omicron RBDs were tighter than its affinity for the WT RBD, with BA.1 RBD binding ~1.7-fold tighter and the other Omicron RBD constructs tested binding ~4-5.5-fold tighter relative to WT RBD.
Omicron RBD に対する ACE2 親和性は WT RBD に対する親和性よりも強く、BA.1 RBD の結合は約 1.7 倍強く、試験した他の Omicron RBD コンストラクトの結合は WT RBD と比較して約 4 ~ 5.5 倍強くなりました。
For variants where both the RBD and S ectodomians were tested, enhanced ACE2 affinities was observed for the RBD constructs than for the corresponding S proteins, with the fold change in RBD affinity with respect to S ectodomains affinity being higher for the Omicron XBB.1.5, XBB.1.16 and EG.5 variants compared to the D614G variant (Figure 2I-J).
RBD と S 外腺細胞の両方をテストしたバリアントでは、対応する S タンパク質よりも RBD 構築物の ACE2 親和性の向上が観察されました。 S外部ドメイン親和性に対するRBD親和性の変化倍数は、D614G変異体と比較して、Omicron XBB.1.5、XBB.1.16およびEG.5変異体の方が高かった(図2I~J)。
Higher association rates(kon) were primarily responsible for the enhanced ACE2 affinities in the RBD only constructs, consistent with a reduction in productive ACE2-RBM encounters when the RBD is part of the S protein.
より高い結合率(kon)は、RBD のみの構築物における ACE2 親和性の向上に主に関与しており、RBD が S タンパク質の一部である場合の生産的な ACE2-RBM 遭遇の減少と一致しています。
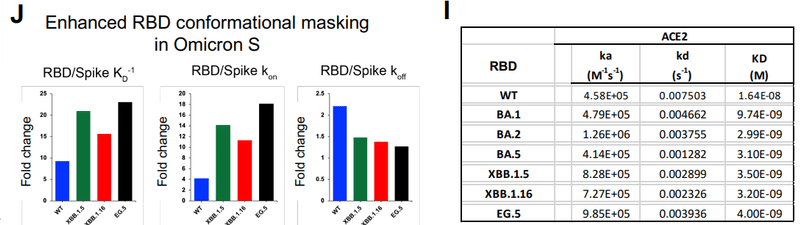
The differences in ACE2 binding affinity and kinetics between the RBD-only construct and the S ectodomains are a measure of the conformational masking that the RBM is subjected to in the context of the S protein.
RBD のみの構築物と S 外部ドメインの間の ACE2 結合親和性と反応速度の違いは、S タンパク質との関連で RBM が受ける立体構造マスキングの尺度です。
Our data demonstrate that conformational masking and shielding of the RBM is greater for the Omicron RBDs than the WT RBD.
我々のデータは、RBM の立体構造マスキングとシールドが WT RBD よりも Omicron RBD の方が優れていることを示しています。
Thus, the increased ACE2 binding affinity of the Omicron S proteins relative to the D614G S protein (Figure 2G) was achieved by the increased intrinsic affinity of ACE2 for the Omicron RBDs counteracting the enhanced conformational masking of the RBM in the Omicron S proteins (Figure 2H-J).
したがって、 D614G S タンパク質と比較した Omicron S タンパク質の ACE2 結合親和性の増加(図 2G)は、Omicron S タンパク質における RBM の立体構造マスキングの強化に対抗する、Omicron RBD に対する ACE2 の固有の親和性の増加によって達成されました (図 2H ~ J)。
Cryo-EM structures of XBB lineage S ectodomains
XBB 系統の細胞外部ドメインのクライオ EM 構造
To visualize their structures and conformations, we determined cryo-EM structures of S protein ectodomains of XBB.1.5, XBB.1.16, EG.5 and EG.5.1 (Figures 3-5, Data S3-S4 and Table S1), utilizing our S-GSAS platform (Figure S2 and Data S1).
それらの構造と立体構造を視覚化するために、XBB.1.5、XBB.1.16、EG.5、および EG.5.1 の S タンパク質細胞外ドメインの低温 EM 構造を決定しました (図 3-5、データ S3-S4 および表 S1)。我々の S-GSAS プラットフォームを利用します (図 S2 およびデータ S1)。
For XBB.1.16, we also determined the structure of S-RRAR-XBB.1.16, a construct that contained the native furin cleavage site between the S1 and S2 subunits, with the S protein cleaved with furin during protein expression (Figures 4, S2, Data S1, S3, S4 and Table S1).
XBB.1.16 については、S-RRAR-XBB.1.16 の構造も決定しました。この構築物は、S1 サブユニットと S2 サブユニットの間に天然のフューリン切断部位を含み、タンパク質発現中に S タンパク質がフューリンで切断される構造です (図 4、S2、データ S1、S3、S4、および表 S1)。
Cryo-EM structures of the XBB.1.5 S protein ectodomain
XBB.1.5 S タンパク質外部ドメインのクライオ EM 構造
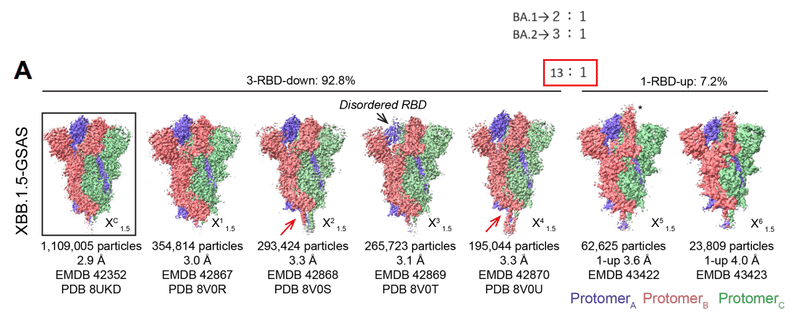
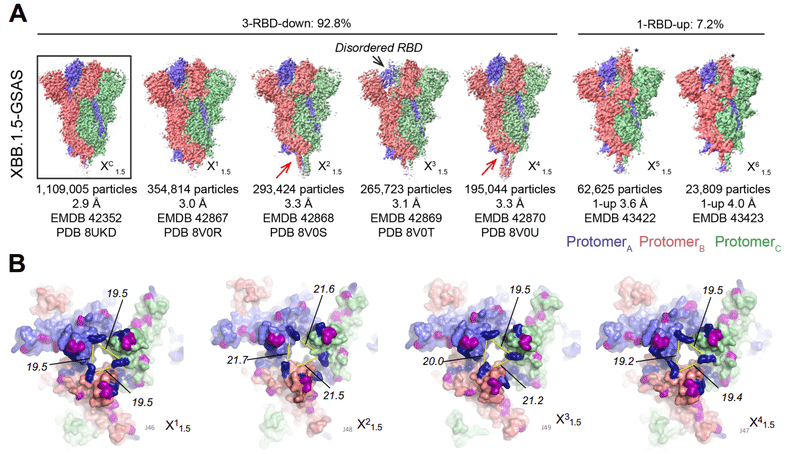
For S-GSAS-XBB.1.5 (Figure 3, Data S3-S4 and Table S1), we identified 3-RBD-down (closed) and 1-RBD-up (open) S ectodomain particle populations in a roughly 13:1 ratio of closed to open, which was substantially higher than the 3:1 and 2:1 ratios of closed to open S we had observed for BA.2 S and BA.1 S, respectively.
S-GSAS-XBB.1.5 (図 3、データ S3 ~ S4 および表 S1) については、私たちは、3-RBD-down (閉じた) および 1-RBD-up (開いた) 外部ドメイン粒子集団を、閉じたものと開いたものの比率がおよそ 13:1 であることを確認しました。 これは、BA.2 S と BA.1 S でそれぞれ観察されたクローズド S とオープン S の比率 3:1 および 2:1 よりも大幅に高かった。
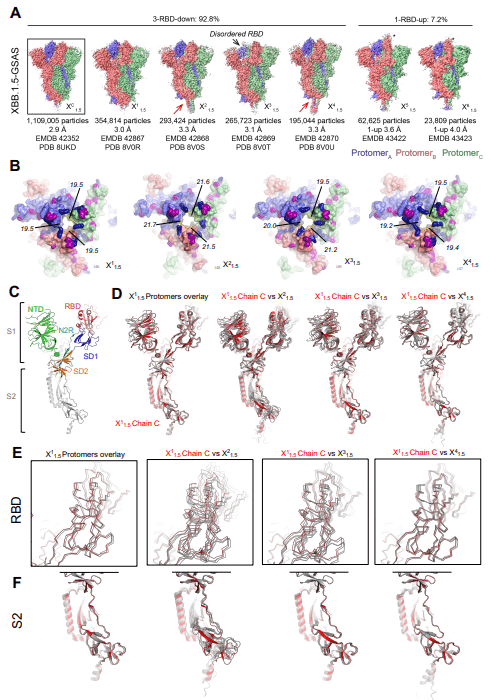
The dominant 3-RBD-down population yielded a 2.9 Å reconstruction, named XDC1.5 here (PDB: 8UKD; EMDB: 42352) (Figure 3A).
優勢な 3-RBD-down 集団は、ここでは XDC1.5 と名付けられた 2.9 Å の再構築をもたらしました (PDB: 8UKD; EMDB: 42352) (図 3A)。
Upon further subclassification, we obtained 4 and 2 classes, respectively, for the 3-RBD-down and 1-RBD-up populations.
さらに細分類すると、3-RBD-down 集団と 1-RBD-up 集団についてそれぞれ 4 クラスと 2 クラスが得られました。
These were designated X11.5 (PDB: 8V0R, EMDB: 42867), X21.5 (PDB: 8V0S, EMDB: 42868), X31.5 (PDB: 8V0T, EMDB: 42869), X41.5 (PDB: 8V0U, EMDB: 42870), X51.5 (EMDB: 43422), X61.5 (EMDB: 43423).
これらは X1-1.5 (PDB: 8V0R、EMDB: 42867) 、X2-1.5 (PDB: 8V0S、EMDB: 42868)、X3-1.5 (PDB: 8V0T、EMDB: 42869)、X4-1.5 (PDB: 8V0U、EMDB: 42870)、X5-1.5 (EMDB: 43422)、X6-1. 5 (EMDB: 43423)と指定されました。
In a study reporting the cryo-EM structural determination of XBB.1 S protein, only the 3-RBD-down, closed state of the S protein was identified in the cryo-EM dataset, suggesting a strong propensity for adopting the 3-RBD-down closed state.
XBB.1 S タンパク質の低温 EM 構造決定を報告する研究では、S タンパク質の 3-RBD ダウンの閉じた状態のみが低温 EM データセットで特定されました。これは、3-RBD-down 閉鎖状態を採用する強い傾向を示唆しています。
Indeed, in this study initially the cryo-EM data analysis only yielded the 3-RBD-down state for the XBB.1.5 S ectodomain cryo-EM dataset, with the 1-RBD-up state being revealed upon extensive classification.
実際、この研究では当初、クライオ EM データ分析では XBB.1.5 外部ドメインのクライオ EM データセットの 3-RBD-down 状態のみが得られ、1-RBD-up 状態は広範な分類によって明らかになりました。
The consensus 1-RBD-up population XUC1.5 showed a highly disordered up RBD characterized by missing or weak density (Figure S4).
コンセンサス 1-RBD-up 集団 XUC1.5 は、密度が欠落しているか密度が低いことを特徴とする高度に無秩序な up RBD を示しました (図 S4)。
Upon further classification to resolve disorder of the XUC1.5 population, two subclasses, named X51.5 and X61.5 here, were revealed that differed primarily by the position of the up-RBD, with the RBD in X61.5 adopting an intermediate conformation leaning more towards the central trimer axis and retaining contact with the RBD and NTD from the two adjacent protomers, while in X51.5 the up RBD adopted a more erect conformation leaning away from the central trimer axis in one direction, and leaning sideways in an orthogonal direction towards the adjacent RBD and away from the adjacent NTD (Figures 3A and S4).
XUC1.5 集団の障害を解決するためにさらに分類すると、ここでは X5-1.5 および X6-1.5 と名付けられた 2 つのサブクラスが、主に上向き RBD の位置によって異なることが明らかになりました。X6-1.5 の RBD は、中心三量体軸により傾いた中間構造を採用し、隣接する 2 つのプロトマーからの RBD および NTD との接触を維持します。X5-1.5 では、アップ RBD は三量体の中心軸から一方向に傾いたより直立した構造を採用しました。そして、隣接する RBD に向かって、隣接する NTD から遠ざかる直交方向に横に傾いています(図 3A および S4)。
Thus, the disordered or missing “up” RBD density in the consensus structure was a result of flexibility, with the RBD adopting varying dispositions around the canonical “up” configuration within the S protein molecules in the population.
したがって、コンセンサス構造における無秩序または欠落した「アップ」RBD 密度は、柔軟性の結果でした。RBD は、集団内の S タンパク質分子内の標準的な「アップ」配置を中心にさまざまな性質を採用しています。
Clear differences were discreible between the four XBB.1.5 3-RBD-down S protein subclasses (Figure 3A-B).
4 つの XBB.1.5 3-RBD-down S タンパク質サブクラス間では、明らかな違いが識別できました (図 3A ~ B)。
The X11.5 subclass consisted of the largest number of particles and most closely resembled the consensus structure XDC1.5 with an overall 0.26 Å C alpha root mean square deviation (Cα RMSD) between the two structures.
X1-1.5 サブクラスは最大数の粒子で構成され、コンセンサス構造 XDC1.5 に最もよく似ていました。2 つの構造間の全体的な C アルファ二乗平均平方根偏差 (Cα RMSD) は 0.26 Å です。
The most divergent 3-RBD-down subclass was X21.5 with overall 2.16 Å and 2.26 Å Cα RMSD, relative to XDC1.5 and X11.5, respectively.
最も多様な 3-RBD-down サブクラスは X2-1.5 で、XDC1.5 および X1-1.5 と比較して、全体の Cα RMSD がそれぞれ 2.16 Å および 2.26 Å でした。
The two other 3-RBD-down subpopulations X31.5 and X41.5 exhibited overall Cα RMSD of 0.32 Å and 0.37 Å, respectively, relative to the consensus XDC1.5, and 0.19 Å and 0.22 Å, respectively, relative to the X11.5 subclass.
他の 2 つの 3-RBD-down 亜集団 X3-1.5 および X4-1.5 は、コンセンサス XDC1.5 と比較して、それぞれ 0.32 Å および 0.37 Å の全体的な Cα RMSD を示しました。X1-1.5 サブクラスと比較した場合は、それぞれ 0.19 Å と 0.22 Å です。
Although not reflected in their overall Cα RMSDs, local differences were observed which distinguished the X31.5 and X41.5 populations from each other and from the other subclasses.
全体的な Cα RMSD には反映されていませんが、X3-1.5 集団と X4-1.5 集団を相互に、また他のサブクラスと区別する局所的な差異が観察されました。
In X31.5, the cryo-EM map density for one of the three RBDs appeared weaker than the other two, suggesting variability in the RBD position.
X3-1.5 では、3 つの RBD のうち 1 つのクライオ EM マップ密度が他の 2 つよりも弱く見えました。RBD の位置にばらつきがあることを示唆しています。
The residues at the C-termini in X21.5 and X41.5 were more ordered than they typically are in S ectodomain structures and were hinged at an angle relative to the body of the spike in a manner reminiscent of the hinging motion described in cryo-ET structures of the SARS-CoV-2 spike.
X2-1.5 および X4-1.5 の C 末端の残基は、通常の外部ドメイン構造よりも規則的でした。これらは、SARS-CoV-2 スパイクのクライオ ET 構造で説明されているヒンジ動作を彷彿とさせる方法で、スパイクの本体に対してある角度でヒンジで取り付けられていました。
A notable difference observed between the 3-RBD-down subclasses were in the relative disposition of their RBDs, with the RBDs of two of the subclasses X11.5 and X41.5 more compactly packed than of the two others (X21.5 and X31.5) (Figure 3B).
3-RBD-down サブクラス間で観察された顕著な違いは、RBD の相対的性質にありました。サブクラス X1-1.5 および X4-1.5 の 2 つの RBD は、他の 2 つのサブクラス (X2-1.5 および X3-1.5) よりもコンパクトにパックされています (図 3B)。
The separation between the RBDs was quantified by measuring the distance between the Cα atoms of residue K440 in the different protomers of each structure.
RBD間の分離は、各構造の異なるプロトマーにおける残基K440のCα原子間の距離を測定することによって定量化された。
All three distances were 19.5 Å for X11.5, and between 19.2 to19.5 Å for X41.5.
X1-1.5 では 3 つの距離すべてが 19.5 Å でした。X4-1.5 の場合は 19.2 ~ 19.5 Å です。
These distances measured between 21.5 to 21.7 Å for X21.5 and between 19.5 to 21.2 Å for X31.5.
これらの距離は、X2-1.5 で 21.5 ~ 21.7 Å の間で測定されました。 X3-1.5 の場合は 19.5 ~ 21.2 Å です。
The three protomers of X11.5 overlaid by their SD2 subdomains showed good overlap throughout except for expected minor variations in the NTD region (Figure 3C and D), indicating a near symmetric structure despite no symmetry having been applied during the reconstructions.
SD2 サブドメインが重なった X1-1.5 の 3 つのプロトマーは、NTD 領域で予想されるわずかな変化を除いて、全体にわたって良好な重複を示しました (図 3C および D)。これは、再構成中に対称性が適用されていないにもかかわらず、ほぼ対称的な構造を示しています。
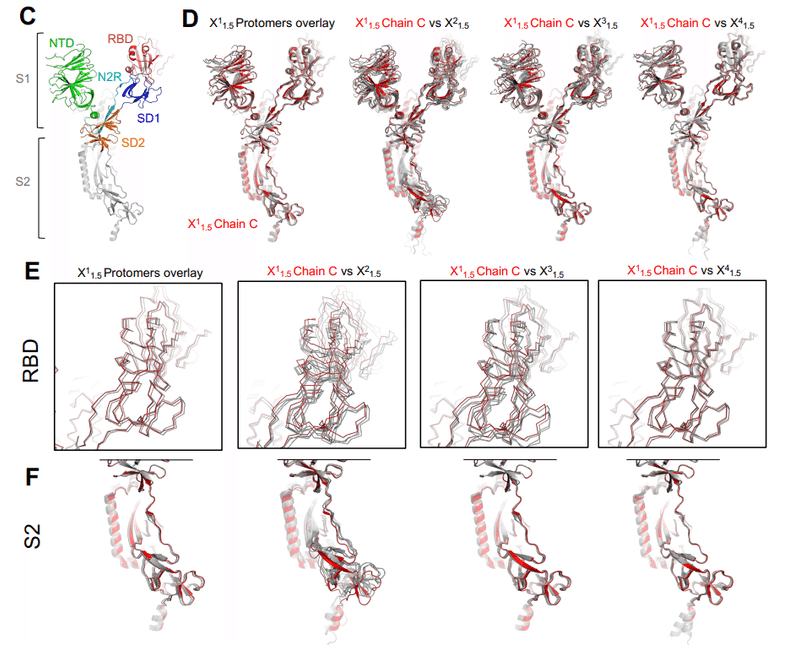
We next overlaid one of X11.5 protomers with the three protomers of X21.5, X31.5 and X41.5, respectively.
次に、X1-1.5 プロトマーの 1 つを、それぞれ X2-1.5、X3-1.5、および X4-1.5 の 3 つのプロトマーとオーバーレイしました。
The RBD of the three X41.5 protomers overlaid well with the RBD of X11.5 (Figure 3D and E).
3 つの X4-1.5 プロトマーの RBD は、X1-1.5 の RBD とよく重なっていました (図 3D および E)。
The highest divergence of RBD positions between protomers was seen for X21.5, followed by X31.5.
プロトマー間の RBD 位置の最も大きな相違は X2-1.5 で見られ、次に X3-1.5 が続きました。
For X31.5, although the RBD positions diverged, the S2 regions of the different protomers overlaid well with each other and with the S2 of X11.5, while for the X21.5 structure, differences in the S2 regions were visible throughout, both between its own protomers and with the X11.5 protomer.
X3-1.5 では、RBD の位置は分岐していましたが、異なるプロトマーの S2 領域は相互に、また X1-1.5 の S2 とよく重なっていました。一方、X2-1.5 構造では、それ自体のプロトマー間および X1-1.5 プロトマーとの両方で、S2 領域の違いが全体にわたって見られました。
In summary, the XBB.1.5 S ectodomain cryo-EM dataset revealed considerable diversity in its dominant 3-RBD-down population with conformational variability observed in RBD dispositions as well as in other regions of the S protein, including in the typically invariant S2 subunit.
要約すると、XBB.1.5 外部ドメインのクライオ EM データセットは、RBD の性質に観察される立体構造の多様性を伴う、その優勢な 3-RBD ダウン集団のかなりの多様性を明らかにしました。 同様に、典型的には不変の S2 サブユニットを含む S タンパク質の他の領域にも多様性は存在します。
Cryo-EM structures of the XBB.1.16 S protein ectodomain
XBB.1.16 S タンパク質外部ドメインのクライオ EM 構造
The XBB.1.16 S protein differs from the XBB.1.5 S protein by two residue substitutions, E180V in the NTD and K478R in the RBD (Figures 1A and S2).
XBB.1.16 S タンパク質は、NTD の E180V と RBD の K478R という 2 つの残基置換によって XBB.1.5 S タンパク質と異なります (図 1A および S2)。
The baseline forms of XBB.1.5 and XBB.1.16 showed similar ability to evade humoral immunity and therapeutic antibodies as did XBB.1, although both showed increased infectivity in pseudovirus assays compared to XBB.1, with XBB.1.16 demonstrating greater growth advantage in the human population compared to XBB.1 and XBB.1.5.
XBB.1.5 および XBB.1.16 のベースライン フォームは、XBB.1 と同様に体液性免疫および治療用抗体を回避する能力を示しました。ただし、両方とも XBB.1 と比較してシュードウイルス アッセイにおいて感染力の増加を示し、XBB.1.16 は XBB.1 および XBB.1.5 と比較してヒト集団におけるより大きな増殖利点を示しました。
In the S-GSAS-XBB.1.16 cryo-EM dataset (Figure 4, Data S3-S4, Table S1), we identified 3-RBD-down and 1-RBD-up S populations in a ∼9:1 ratio.
S-GSAS-XBB.1.16 クライオ EM データセット (図 4、データ S3 ~ S4、表 S1) では、3-RBD-down と 1-RBD-up S 集団が約 9:1 の比率で特定されました。
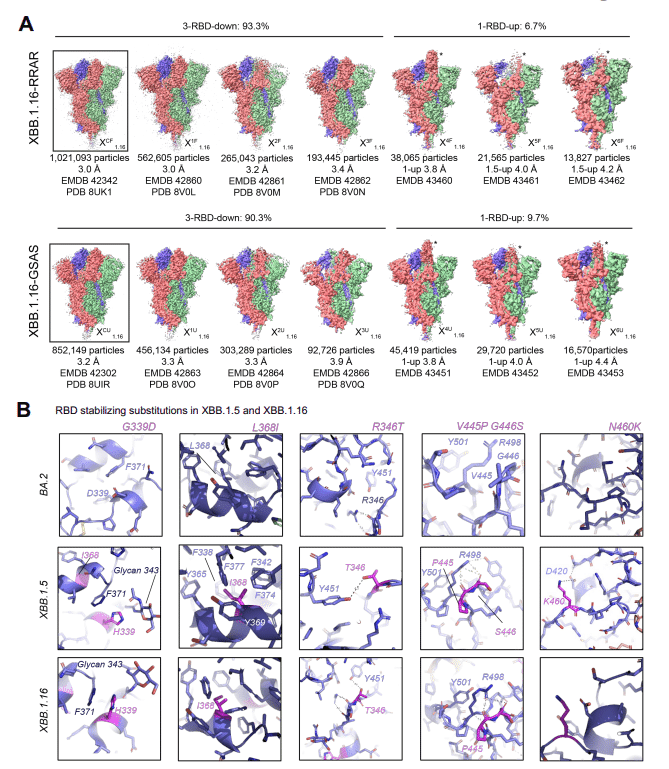
We also determined the structures of the furin-cleaved S-RRAR-XBB.1.16 construct, where we observed ∼14:1 ratio of closed to open S conformations.
また、フーリンで切断された S-RRAR-XBB.1.16 コンストラクトの構造も決定し、閉じた S 構造と開いた S 構造の比率が約 14:1 であることが観察されました。
The 1-RBD-up population of the uncleaved S-GSAS-XBB.1.16 S ectodomain showed expected variability in the disposition of the up RBD that was sampled in three subclasses, named X41.16, X51.16 and X61.16.
切断されていない S-GSAS-XBB.1.16 外部ドメインの 1-RBD-up 集団は、up RBD の性質に予想される変動性を示しました。これは、X4-1.16、X5-1.16、および X6-1.16 という名前の 3 つのサブクラスでサンプリングされました。
In the furin-cleaved S-RRAR-XBB.1.16 S ectodomain cryo-EM dataset, we identified not only 1-RBD-up S ectodomains but also populations where a second RBD appeared partially in the up position suggesting that the furin-cleaved S protein may be more flexible in the RBD-up or open states than the uncleaved XBB.1.16 S protein.
フューリンで切断された S-RRAR-XBB.1.16 細胞外部ドメインのクライオ EM データセットでは、私たちは、1-RBD-up S細胞外部ドメインだけでなく、2番目のRBDが部分的に上の位置に出現する集団も特定しました。これは、フューリンで切断された S タンパク質が、RBD アップまたはオープン状態において、切断されていない XBB.1.16 S タンパク質よりも柔軟性がある可能性があることを示唆しています。
The 3-RBD-down structures of the uncleaved and furin-cleaved versions of the XBB.1.16 S ectodomain were similar with overall Cα RMSD of 0.3 Å.
XBB.1.16 S外部ドメインの未切断バージョンとフリン切断バージョンの3-RBDダウン構造は類似しており、全体のCα RMSDは0.3 Åでした。
The 3-RBD-down population were subclassified into three populations each for the uncleaved and furin cleaved datasets of the XBB.1.16 S ectodomain.
3-RBD-down 集団は、XBB.1.16 S 外部ドメインの非切断データセットとフリン切断データセットごとに 3 つの集団に下位分類されました。
The populations were more homogeneous and the differences between the subclasses subtle compared to what was observed for the XBB.1.5 S protein, with overall Cα RMSDs between all the XBB.1.16 stuctures being in the 0.20-0.40 Å range.
XBB.1.5 S タンパク質で観察されたものと比較すると、集団はより均質であり、サブクラス間の差異はわずかでした。すべての XBB.1.16 構造間の全体的な Cα RMSD は 0.20 ~ 0.40 Å の範囲にあります。
Several residue substitutions in the XBB.1.5 and XBB.1.16 S proteins occurred at the ACE2 binding site (Figure 1A), including a rare F486P substitution that involved a two-nucleotide mutation and was implicated in the phenotypic change in XBB.1.5 that resulted in its gaining dominance over XBB.1.
XBB.1.5 および XBB.1.16 S タンパク質のいくつかの残基置換は、ACE2 結合部位で発生しました (図 1A)。これには、2 ヌクレオチドの変異が関与し、XBB.1.1 よりも優位性を獲得した XBB.1.5 の表現型変化に関与する稀な F486P 置換が含まれます。
The F486P substitution facilitated enhanced ACE2 binding, greater transmissibility and escape from neutralizing antibodies.
F486P 置換により、ACE2 結合が強化され、伝達性が高まり、中和抗体からの回避が促進されました。
F490S, another substitution in the RBM (residues 483–506), observed previously in the Delta VOC and Lambda VOI, was implicated in resistance to neutralizing antibodies.
デルタ VOC およびラムダ VOI で以前に観察された RBM の別の置換 (残基 483 ~ 506) である F490S は、中和抗体に対する耐性に関与していました。
For XBB.1.5 and XBB.1.16 S ectodomains, we observed the RBD-RBD down state packing that we have previously reported for the BA.2 S protein (Figure S5A).
XBB.1.5 および XBB.1.16 S 外部ドメインについては、BA.2 S タンパク質について以前に報告した RBD-RBD ダウン状態パッキングを観察しました (図 S5A)。
No additional mutations were acquired at the RBD-RBD down state interprotomer contact sites of the XBB.1.5 and XBB.1.16 S proteins, and none of the previously acquired mutations at these sites were reverted or changed (Figure 1A-B).
XBB.1.5 および XBB.1.16 S タンパク質の RBD-RBD ダウン状態プロトマー間接触部位では追加の変異は獲得されず、これらの部位で以前に獲得された変異はいずれも復帰または変化しませんでした (図 1A-B)。
We found that the new RBD mutations either improved immune evasion by targeting the most immunodominant sites on the RBD or they facilitated interactions within the RBD, thereby improving RBD stability (Figures 1A-C).
私たちは、新しい RBD 変異が RBD 上の最も免疫優勢な部位を標的とすることで免疫回避を改善するか、または RBD 内の相互作用を促進することを発見しました。これにより、RBD の安定性が向上します (図 1A ~ C)。
The Omicron BA.1 and BA.2 clades both carried a G339D substitution, and an additional S371F emerged in BA.2 that was preserved in subsequent linages.
Omicron BA.1 クレードと BA.2 クレードはどちらも G339D 置換を有しており、BA.2 では追加の S371F が出現し、それが後続の系統で保存されました。
The G339D substitution is located at a site of intraprotomer RBD stabilization with the BA.2 S371F substitution facilitating close packing of two intra-RBD helical stretches 339–342 and 367–371 (Figure 4B).
G339D 置換はプロトマー内 RBD 安定化部位に位置しており、BA.2 S371F 置換は 2 つの RBD 内のらせんストレッチ 339 ~ 342 および 367 ~ 371 の密な充填を促進します (図 4B)。
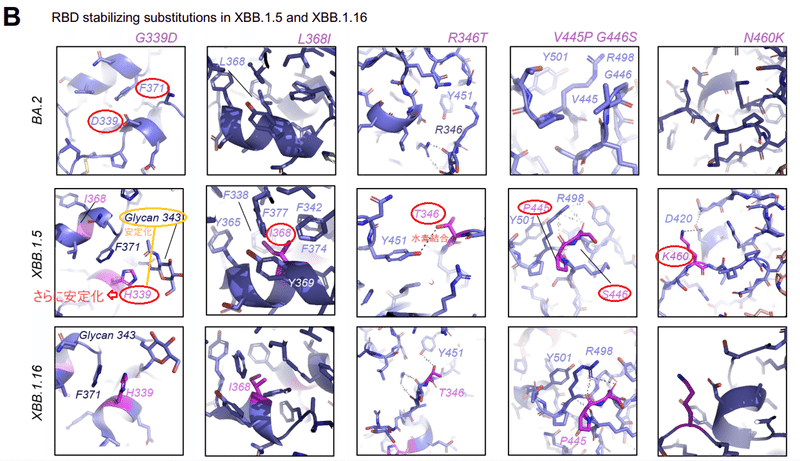
In XBB.1.5 a histidine appeared at residue 339 and further stabilized this region by packing against the side chain of F371 via a π-π stacking interaction, thus highlighting this region as a hotspot for RBD stabilization.
XBB.1.5 では、ヒスチジンが残基 339 に出現し、π-π スタッキング相互作用を介して F371 の側鎖に対してパッキングすることでこの領域をさらに安定化しました。したがって、この領域が RBD 安定化のホットスポットとして強調されます。
Additionally, H339 was positioned to stack against and hydrogen bond with residue N343 and the N343 glycan, enhancing intra-helical stability and stabilizing and orienting glycan 343.
さらに、H339 は残基 N343 および N343 グリカンに対して積み重ねて水素結合するように配置され、ヘリックス内の安定性を高め、グリカン 343 を安定化および配向させました。
Furthermore, the L368I substitution that appears in XBB.1.5 occurs within the 367-371 helical stretch at the center of an aromatic/hydrophobic cluster involving residues F371, Y365, F377, F374, F342, F338 and V367.
さらに、XBB.1.5 に見られる L368I 置換は、残基 F371、Y365、F377、F374、F342、F338、および V367 を含む芳香族/疎水性クラスターの中心の 367-371 螺旋ストレッチ内で発生します。
Thus, it appears that the regions around the S371F, G339H and L368I are being optimized as Omicron evolves to stabilize the RBD while it engages in interprotomer down state RBD-RBD contacts.
したがって、Omicronがプロトマーダウン状態のRBD-RBD接触を行いながらRBDを安定化するように進化するにつれて、S371F、G339H、およびL368Iの周囲の領域が最適化されているようです。
Other stabilizing mutations in XBB.1.5 and XBB.1.16 include a R346T substitution in a RBD surface loop, a substitution that appeared in multiple lineages, including some that expanded, e.g. BQ.1.1, BA.4.6, BA.5.2.6, XBF, and FU.1.
XBB.1.5 および XBB.1.16 の他の安定化変異には、RBD 表面ループの R346T 置換が含まれます。 これには、BQ.1.1、BA.4.6、BA.5.2.6、XBF、FU.1 などの拡張された複数の系統にも現れました。
Shortening the side chain from an Arginine to a Threonine at position 346 brings the hydroxyl group within hydrogen bonding distance to the Y451 side chain hydroxyl (Figure 4B).
346 位の側鎖をアルギニンからスレオニンに短くすると、ヒドロキシル基が Y451 側鎖ヒドロキシルとの水素結合距離内に収まります (図 4B)。
A substitution of two consecutive residues V455P and G446S occurred in XBB.1.5 and XBB.1.16, where G446S was acquired in BA.1, reverted in BA.2 to G and changed to S again in XBB.1.5.
XBB.1.5 と XBB.1.16 では、2 つの連続する残基 V455P と G446S の置換が発生しました。ここで、G446S は BA.1 で取得され、BA.2 で G に戻り、XBB.1.5 で再び S に変更されました。
Together they stabilize a turn in a surface loop that packs against residue R498 that we had previously shown forms a π-cation pair with Y501, both residues acquired in Omicron BA.1 via the Q498R and N501Y substitutions and retained in later variants.
これらは一緒になって、Y501とπカチオンペアを形成することを以前に示した残基R498に対してパックする表面ループのターンを安定化します。どちらの残基も Q498R および N501Y 置換を介して Omicron BA.1 に取得され、後のバリアントにも保持されています。
The R498-Y501 pair stabilizes a loop that contributes to interprotomer stabilizing contacts between down state RBDs.
R498-Y501 ペアは、ダウン状態の RBD 間のプロトマー間接触の安定化に寄与するループを安定化します。
The N460K substitution occurs in a surface loop and forms a salt bridge with D420.
N460K 置換は表面ループで発生し、D420 と塩橋を形成します。
In summary, the XBB lineage incorporated RBD substitutions that improved RBD stability while also maintaining the dominance of the 3-RBD-down state, which likely contributes to its immune evasive properties by conformational masking of the immunodominant RBD regions.
要約すると、XBB 系統には、3-RBD ダウン状態の優位性を維持しながら RBD の安定性を向上させる RBD 置換が組み込まれています。これは、免疫優勢な RBD 領域の構造的マスキングによってその免疫回避特性に寄与している可能性があります。
Cryo-EM structures of EG.5 and EG.5.1 S ectodomains
EG.5 および EG.5.1 細胞外ドメインのクライオ EM 構造
To visualize the structures and conformations of the EG.5 S protein, we obtained a cryo-EM dataset of S-GSAS-EG.5 (Figures 5, Data S3-S4 and Table S1).
EG.5 S タンパク質の構造と立体構造を視覚化するために、S-GSAS-EG.5 のクライオ EM データセットを取得しました (図 5、データ S3 ~ S4 および表 S1)。
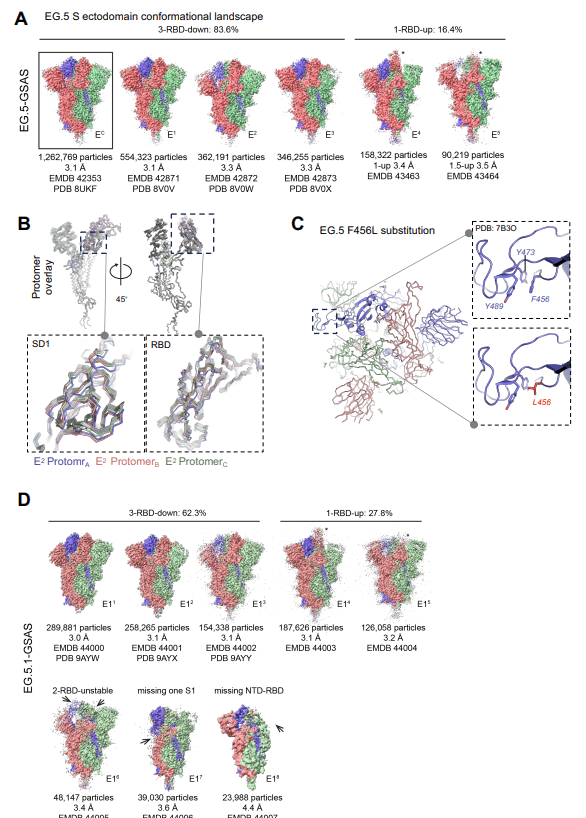
EG.5 has a single F456L substitution relative to XBB.1.5 located in the RBD (Figure 1A and S2).
EG.5 には、RBD に位置する XBB.1.5 と比較して F456L 置換が 1 つあります (図 1A および S2)。
The 3-RBD-down population still dominated in the EG.5 S cryo-EM dataset but its proportion relative the RBD-up populations (∼5:1) was smaller compared to what was observed in the XBB.1.5 and XBB.1.16 S ectodomain cryo-EM datasets.
3-RBD-down 集団は依然として EG.5 S クライオ EM データセットで優勢ですが、RBD-up 集団に対するその割合 (約 5:1) は、XBB.1.5 および XBB.1.16 外部ドメインのクライオ EM データセットで観察されたものと比較して小さかった。
The EG.5 S protein 3-RBD-down consensus structure EC (PDB: 8UKF; EMDB: 42353), was subclassified into E1 (PDB: 8VOV; EMDB: 42871), E2 (PDB: 8VOW; EMDB: 42872) and E3 (PDB: 8VOX; EMDB: 42873).
EG.5 S タンパク質 3-RBD ダウン コンセンサス構造 EC (PDB: 8UKF; EMDB: 42353) は、E1 (PDB: 8VOV; EMDB: 42871)、E2 (PDB: 8VOW; EMDB: 42872)、および E3 (PDB: 8VOX; EMDB: 42873) に下位分類されました。
Considerable variability between the 3-RBD-down subclasses was observed in the S1 subunit, with E1 being the most ordered with well-packed RBDs and E2 the least (Figure S5B).
3-RBD-down サブクラス間のかなりのばらつきが S1 サブユニットで観察されました。E1 は十分に詰め込まれた RBD で最も順序付けられており、E2 は最も小さくなっています (図 S5B)。
The three RBDs in E2 were asymmetrically arranged and showed variable densities with one RBD and its contacting NTD showing the weakest density indicating high disorder.
E2 の 3 つの RBD は非対称に配置され、さまざまな密度を示し、1 つの RBD とそれに接触する NTD は最も弱い密度を示し、高い無秩序性を示しました。
Visualizing the Gaussian filtered map for this reconstruction confirmed that the down state assignment for this RBD.
この再構成のガウス フィルター マップを視覚化すると、この RBD のダウン状態の割り当てが確認されました。
Similarly, one RBD-NTD pair was more disordered than the other two in E3, but less so than in E2.
同様に、E3 では 1 つの RBD-NTD ペアが他の 2 つよりも乱れていましたが、E2 ほど乱れていませんでした。
Comparing the fitted coordinates for the 3-RBD-down structures showed that the subclass with the largest number of particles E1 was the most similar to the consensus structure EC with an overall Cα RMSD of 0.30 Å, followed by E3 and E2 with Cα RMSDs of 0.48 and 0.64 Å, respectively, relative to EC.
3-RBD-down 構造のフィッティング座標を比較すると、最大数の粒子 E1 を持つサブクラスが、全体の Cα RMSD が 0.30 Å のコンセンサス構造 EC に最も類似していることがわかりました。続いて、EC と比較して、Cα RMSD がそれぞれ 0.48 Å および 0.64 Å の E3 と E2 が続きます。
For all the structures, the S2 regions were very similar and overlaid well except at the C-terminal end where some conformational variability was observed (Figure 5B).
すべての構造において、S2 領域は非常に類似しており、C 末端で何らかの立体構造の変動が観察された点を除いて、よく重なっていました (図 5B)。
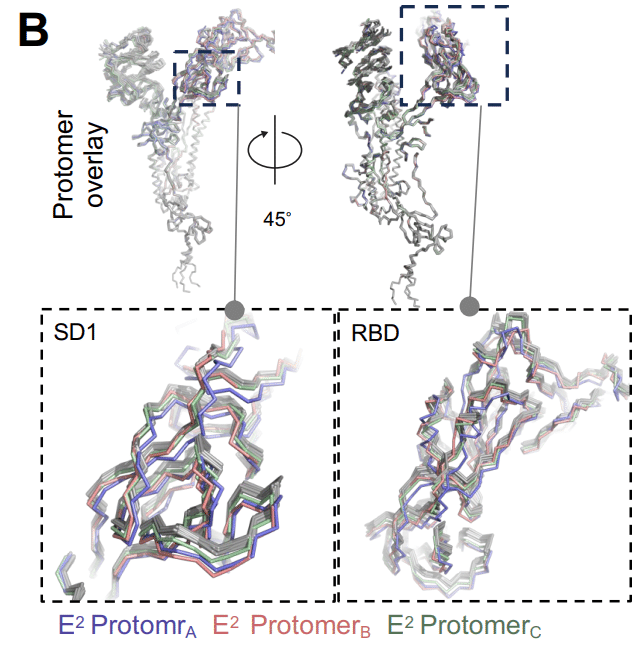
The S1 regions of all except the E2 protomers were similar barring the positional fluctuations in the NTD region that are typically seen in SARS-CoV-2 S protein structures.
E2 プロトマーを除くすべての S1 領域は、SARS-CoV-2 S タンパク質構造で通常見られる NTD 領域の位置変動を除けば、同様でした。
For the E2 structure, two of the protomers showed shifted positions of the SD1 subunit and the RBD (Figure 5B).
E2 構造では、プロトマーのうち 2 つが SD1 サブユニットと RBD の位置のシフトを示しました (図 5B)。
These shifts included the RBD of these protomers tilting away from the central vertical trimer axis, while still in the down position, and this RBD tilting in turn causing changes in the contacting NTD.
これらのシフトには、これらのプロトマーの RBD が中央の垂直三量体軸から離れて傾いていることが含まれます。まだ下位置にある間に、この RBD が傾き、接触している NTD に変化を引き起こします。
Since the resolution of the region around the EG.5 F456L substitution in the EG.5 cryo-EM structures precluded atomic level visualization, we relied on a crystal structure of the RBD (PDB: 7B3O) to understand the structural basis for the effects caused by the F456L substitution (Figure 5C).
EG.5 クライオ EM 構造における EG.5 F456L 置換の周囲の領域の解像度が原子レベルの可視化を妨げたため、私たちは、F456L 置換によって引き起こされる効果の構造的基礎を理解するために、RBD (PDB: 7B3O) の結晶構造に依存しました (図 5C)。
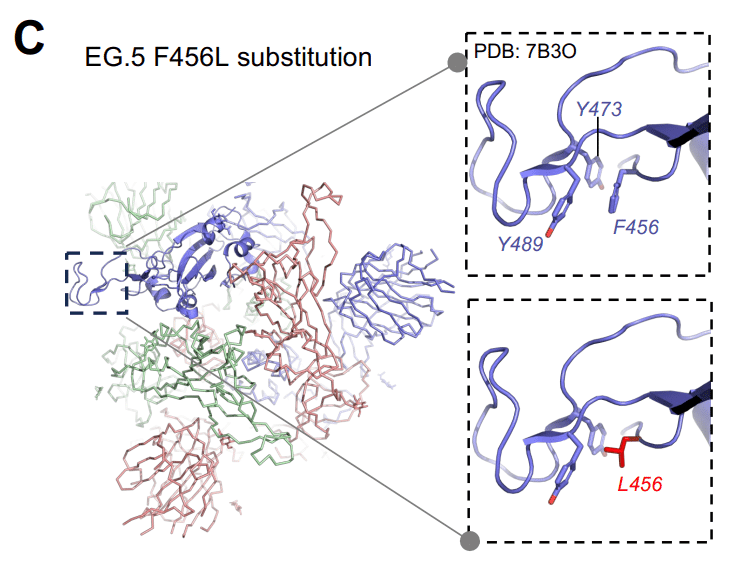
Residue F456 occurs in a region adjacent to the RBD hook that was named previously for its hook-like appearance, where F456 is part of an aromatic cluster with residues Y473 and Y489.
残基F456は、そのフック状の外観から以前に命名されたRBDフックに隣接する領域に存在します。 ここで、F456 は残基 Y473 および Y489 を持つ芳香族クラスターの一部です。
The F456L substitution destabilizes this region by replacing the Phenylalanine with a less bulky, non-aromatic, albeit still hydrophobic residue.
F456L 置換は、依然として疎水性であるにもかかわらず、フェニルアラニンをより嵩張らない非芳香族残基に置き換えることにより、この領域を不安定化します。
Since this region is a key epitope for neutralizing antibodies, the F456L induced structural changes likely contribute to its reported immune evasion properties.
この領域は中和抗体の重要なエピトープであるため、F456L によって誘導される構造変化は、報告されているその免疫回避特性に寄与している可能性があります。
The destabilization of the F456/Y473/Y489 aromatic cluster due to the F456L substitution provides a structural rationale for the reduced thermostability of the EG.5 RBD relative to XBB.1.5 since this mutation is the only differing residue between the S proteins of these variants.
F456L 置換による F456/Y473/Y489 芳香族クラスターの不安定化は、XBB.1.5 と比較して EG.5 RBD の熱安定性が低下する構造的根拠を提供します。この変異は、これらの変異体の S タンパク質間で唯一異なる残基であるためです。
F486L was recurrently selected for within the lineages we studied here subsequent to their initial expansion, indicating that despite its propensity to destabilize the RBD it conferred a selective advantage.
F486L は、ここで研究した系統内で最初の拡大後に繰り返し選択されました。これは、RBD を不安定にする傾向があるにもかかわらず、選択的利点をもたらしたことを示しています。
It was preserved among EG.1 lineages, which originally emerged from within the XBB.1.9 lineage, but was also found independently in the XBB.1.16.6 sublineage, which was beginning to replace XBB.1.16 in North America prior to EG.1’s introduction, and the XBB.1.5 sublineages GK.1 (Pango alias XBB.1.5.70.1) and JD.1 (Pango alias XBB.1.5.102.1) which became dominant lineages in South America just as as EG.1 was expanding in other regions of the world (Fig. S1).
これは、もともと XBB.1.9 系統内から出現した EG.1 系統の間で保存されていましたが、XBB.1.16.6 亜系統でも独立して見つかりました。これは、EG.1 の導入前に北米で XBB.1.16 を置き換え始めていました。XBB.1.5 サブリネージ GK.1 (Pango alias XBB.1.5.70.1) および JD.1 (Pango エイリアス XBB.1.5.102.1) は、EG.1 が世界の他の地域で拡大していたように、南米でも有力なリネージとなりました (図S1)。
EG.5 acquired a single residue substitution Q52H in its NTD to become EG.5.1, the sublineage that spread most rapidly.
EG.5 は、NTD に単一残基置換 Q52H を取得して、最も急速に普及した亜系統である EG.5.1 になりました。
Like EG.5, cryo-EM structures of the EG.5.1 S ectodomain (Figure 5D and S5C) showed heightened S1 subunit disorder, with an increased proportion of RBD up populations, as well small particle populations where parts of the NTD and RBD regions were disordered to an extent that these regions were not visible in the electron density.
EG.5 と同様に、EG.5.1 S 外部ドメインのクライオ EM 構造 (図 5D および S5C) は、S1 サブユニット障害の亢進を示しました。RBD up 集団の割合が増加し、NTD および RBD 領域の一部が電子密度で見えなくなる程度に乱れた小さな粒子集団も発生しました。
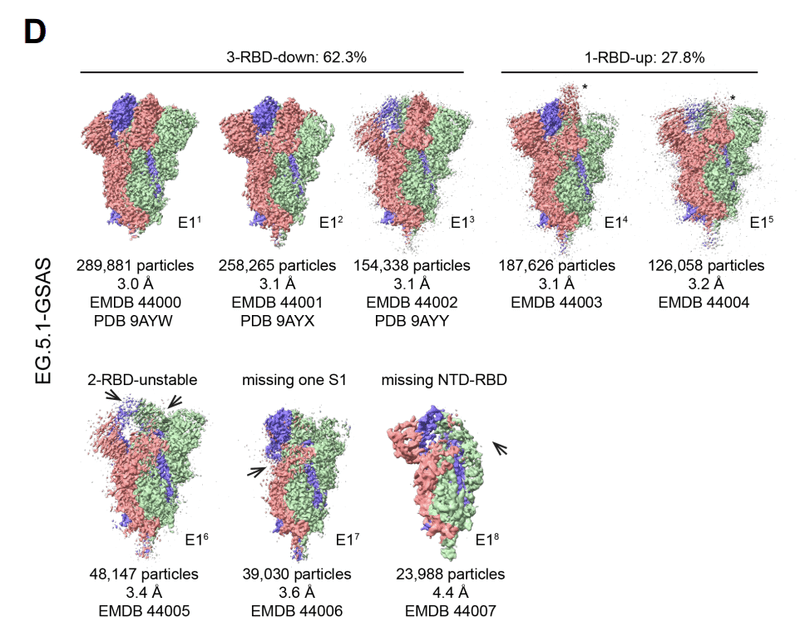
The disordered populations were reminiscent of similarly disordered structures observed in spikes that had transmitted between humans and minks.
この無秩序な個体群は、人間とミンクの間で感染したスパイクで観察された同様の無秩序な構造を彷彿とさせました。
Taken together, our results reveal how substitution of one or two S1 subunit residues can have substantial local and global impacts on S protein structure.
まとめると、我々の結果は、1 つまたは 2 つの S1 サブユニット残基の置換が S タンパク質の構造にどのように局所的および全体的な大きな影響を与える可能性があるかを明らかにします。
Placed in the context of the ongoing evolution of Omicron, our results show that different S protein properties are likely being optimized simultaneously.
Omicron の進行中の進化の文脈に置くと、私たちの結果は、さまざまな S タンパク質の特性が同時に最適化されている可能性が高いことを示しています。
Therefore, despite a trend of increasing RBD stability during Omicron evolution, a single residue substition in EG.5 that destabilized the RBD was nevertheless selected for and retained in the subsequent expansion of the sublineage, likely due to its contribution to immune evasion at an immunodominant site.
したがって、Omicron の進化中に RBD の安定性が増加する傾向があるにもかかわらず、RBD を不安定化する EG.5 の単一残基置換が選択され、その後の亜系統の拡大に保持されました。 おそらく免疫優勢部位での免疫回避への寄与によるものと考えられます。
Antigenicity of the XBB lineage S proteins
XBB系統Sタンパク質の抗原性
To assess the impact of the acquired mutations on antigencity, we tested binding in ELISA to a panel of human anti-S antibodies that bind different regions of the S protein.
抗原性に対する獲得変異の影響を評価するために、S タンパク質のさまざまな領域に結合するヒト抗 S 抗体のパネルへの結合を ELISA でテストしました。
These included S1 antibodies DH1050.1 (NTD-directed), DH1042 (RBD ACE2 binding site-directed), CR3022, DH1047 and S2X259 (RBD inner face directed), and DH1044, DH1193 and S309 (RBD outer face-directed) (Figure 6A-C and S6).
これらには、S1 抗体 DH1050.1 (NTD 指向)、DH1042 (RBD ACE2 結合部位指向)、CR3022、DH1047 および S2X259 (RBD 内面指向)、DH1044、DH1193 および S309 (RBD 外面指向) が含まれます (図6A〜CおよびS6)。
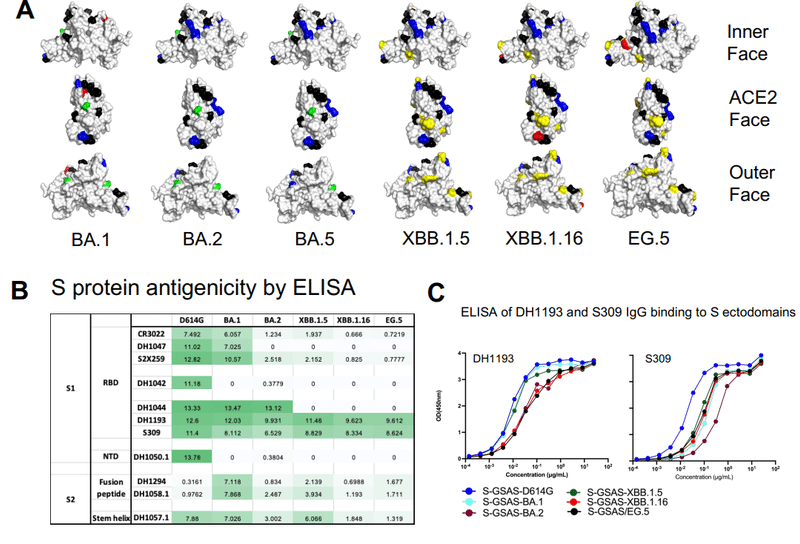
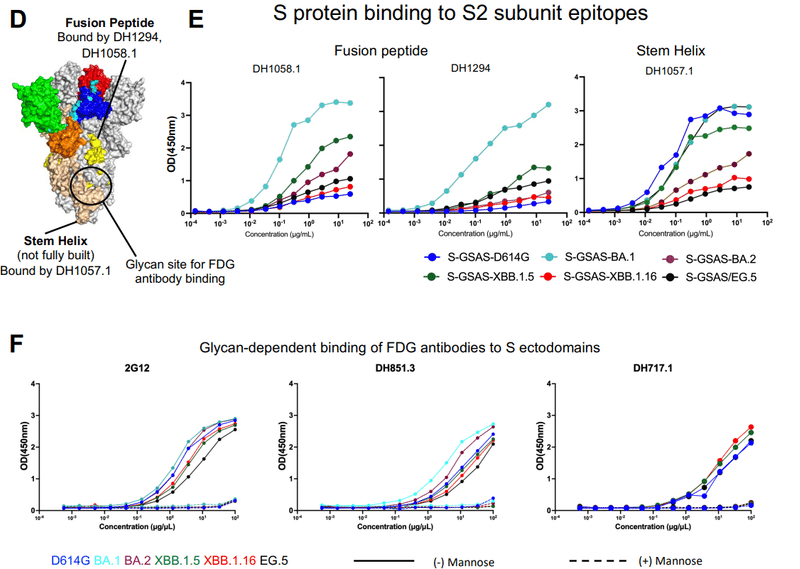
We also included S2 antibodies DH1058.1 and DH1294 (fusion peptide-directed), DH1057.1 (stem helix epitope-directed), and a panel of Fab-dimerized glycan reactive antibodies that target a quaternary cluster of three S2 glycan moieties (Figure 6D, F and S8).
また、S2 抗体 DH1058.1 および DH1294 (融合ペプチド指向)、DH1057.1 (ステムヘリックスエピトープ指向)、および 3 つの S2 グリカン部分の四次クラスターを標的とする Fab 二量体化グリカン反応性抗体のパネルも含めました (図6D、FおよびS8)。
Of all the S1 directed antibodies tested, the only two that showed appreciable binding to the XBB.1.5, XBB.1.16 and EG.5 S ectodomains in ELISA were DH1193 and S309 that bind distinct, albeit overlapping, epitopes on the RBD outer face (Figure 6A-C, S6).
テストされたすべての S1 向け抗体のうち、 ELISA で XBB.1.5、XBB.1.16、および EG.5 S 外部ドメインへのかなりの結合を示した 2 つは、DH1193 と S309 でした。これは、重複しているにもかかわらず、RBD 外面上の別個のエピトープに結合します (図 6A ~ C、S6)。
The ELISA binding of DH1193 and S309 translate to their ability to neutralize in pseudovirus neutralization assays.
DH1193 および S309 の ELISA 結合は、シュードウイルス中和アッセイにおける中和能力に変換されます。
In a recent study, neutralizing antibodies that bound the DH1193 epitope region have been elicited in non-human primates by vaccination, highlighting this region of the RBD as a vaccine target for eliciting neutralizing antibodies that may be less resistant to mutations acquired during SARS-CoV-2 evolution.
最近の研究では、DH1193 エピトープ領域に結合する中和抗体がワクチン接種によって非ヒト霊長類で誘発されています。SARS-CoV-2の進化中に獲得された変異に対する耐性が低い可能性がある中和抗体を誘発するためのワクチン標的として、RBDのこの領域を強調しています。
The EG.5 and EG.5.1 S ectodomains showed similar antigenic properties with robust binding retained to the RBD outer face targeting antibodies S309 and DH1193 (Figure S7).
EG.5 および EG.5.1 S 外部ドメインは、抗体 S309 および DH1193 を標的とする RBD 外面への強い結合が保持され、同様の抗原特性を示しました (図 S7)。
At the S2 subunit, a panel of Fab-dimerized glycan reactive (FDG) antibodies that target a quaternary S2 glycan cluster bound XBB.1.5, XBB.1.16 and EG.5 S ectodomains in a glycan dependent manner (Figure 6F).
S2 サブユニットでは、四次 S2 グリカンクラスターを標的とする Fab 二量体化グリカン反応性 (FDG) 抗体のパネルが、グリカン依存的に XBB.1.5、XBB.1.16、および EG.5 S 外部ドメインに結合しました (図 6F)。
Consistent with previous observations, we found that the furin-cleaved S ectodomains showed stronger binding to the FDG antibodies than the uncleaved versions, demonstrating allosteric effect of furin cleavage on S2 structure and on the presentation of the glycan epitope (Figure S8).
以前の観察と一致して、我々は、フーリンで切断された S エコトドメインが、切断されていないバージョンよりも FDG 抗体に対して強い結合を示すことを発見しました。 S2 構造およびグリカンエピトープの提示に対するフリン切断のアロステリック効果を示しています (図 S8)。
Structural modeling showed that the epitopes of the fusion peptide targeting antibodies DH1058.1 and DH1294, and stem helix targeting antibody DH1057.1, are not fully accessible for antibody binding on the pre-fusion SARS-CoV-2 S protein.
構造モデリングにより、融合ペプチドを標的とする抗体 DH1058.1 および DH1294、およびステムヘリックスを標的とする抗体 DH1057.1 のエピトープは、融合前の SARS-CoV-2 S タンパク質への抗体結合に完全にはアクセスできないことが示されました。
Despite this, measurable binding of the SARS-CoV-2 S protein to these antibodies can be observed by SPR and ELISA, suggesting antibody-induced local refolding of the S protein structure to expose the antibody eptiopes.
それにもかかわらず、SARS-CoV-2 Sタンパク質とこれらの抗体との測定可能な結合はSPRおよびELISAによって観察でき、これは抗体誘導性のSタンパク質構造の局所的なリフォールディングが抗体エピトープを露出させることを示唆しています。
For the fusion peptide-directed antibodies, we have previously reported increased ELISA binding for the BA.1 S ectodomain relative to the D614G S ectodomain, and interpreted this to indicate increased flexibility of this region in the BA.1 S protein leading to greater access to the fusion peptide in the presence of the antibody.
融合ペプチド指向性抗体については、我々は以前に、D614G S 外部ドメインと比較して BA.1 S 外部ドメインに対する ELISA 結合の増加を報告しました。そして、これは、BA.1 Sタンパク質のこの領域の柔軟性が増加し、抗体の存在下で融合ペプチドへのアクセスが向上することを示していると解釈しました。
BA.2 S showed reduced binding to DH1058.1 and DH1294 in ELISA, relative to BA.1 S, indicating stabilization of this region in the pre-fusion S conformation.
BA.2 Sは、ELISAにおいてBA.1 Sと比較してDH1058.1およびDH1294への結合の減少を示し、融合前S立体構造におけるこの領域の安定化を示した。
Our results here recapitulate these trends with BA.1 S showing the highest levels of binding, D614G S the lowest and BA.2 S intermediate between the two (Figure 6E).
ここでの我々の結果は、これらの傾向を再現しており、BA.1 S が最高レベルの結合を示し、D614G S が最低、BA.2 S がその 2 つの中間であることを示しています (図 6E)。
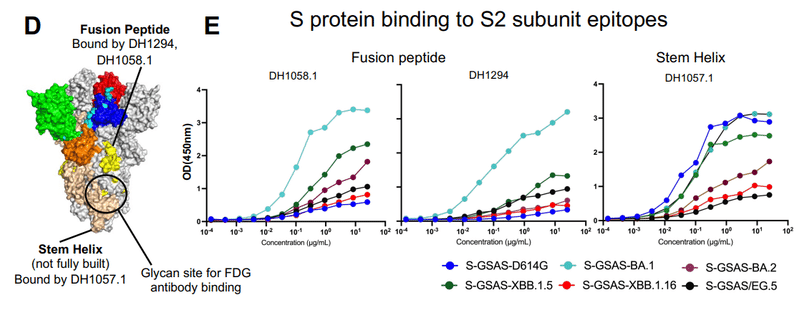
XBB.1.16 and EG.5 S proteins show lower binding to DH1058.1 than BA.2 S, while XBB.1.5 shows higher binding than BA.2 S, although still substantially lower than BA.1 S, demonstrating overall stabilization of this S protein region in its pre-fusion conformation upon Omicron evolution, after being destabilized in the first identified Omicron variant BA.1.
XBB.1.16 および EG.5 S タンパク質は、BA.2 S よりも DH1058.1 への結合が低く、一方、XBB.1.5 は BA.1 S よりも大幅に低いものの、BA.2 S よりも高い結合を示します。この S タンパク質領域は、最初に同定された Omicron 変異体 BA.1 で不安定化された後、Omicron 進化時に融合前立体構造で全体的に安定化することが実証されました。
The difference in ELISA binding observed between XBB.1.5 and XBB.1.16 is notable and consistent with the greater diversity of S2 subunit conformation observed in the XBB.1.5 cryo-EM subpopulations, suggesting greater S2 conformational malleability in the XBB.1.5 S protein than in XBB.1.16 S.
XBB.1.5 と XBB.1.16 の間で観察された ELISA 結合の違いは注目に値し、XBB.1.5 クライオ EM 亜集団で観察された S2 サブユニット構造のより多様性と一致しています。 これは、XBB.1.16 S よりも XBB.1.5 S タンパク質の方が S2 の構造的展性が大きいことを示唆しています。
The second fusion peptide-directed antibody tested DH1294 shows similar trends, although the differences were less pronounced than they were with DH1058.1 (Figure 6E).
2 番目に試験した融合ペプチド指向性抗体 DH1294 も同様の傾向を示しましたが、違いは DH1058.1 ほど顕著ではありませんでした (図 6E)。
The trends observed for DH1057.1 (stem helix) binding was distinct from those observed with the fusion peptide-directed antibodies, with strongest binding observed for the D614G, BA.1 S and XBB.1.5 S ectodomains, and BA.2, XBB.1.16 and EG.5 S ectodomains binding at substantially lower levels (Figure 6E).
DH1057.1 (ステムヘリックス) 結合で観察された傾向は、融合ペプチド指向性抗体で観察された傾向とは異なりました。D614G、BA.1 S、および XBB.1.5 S 外部ドメインでは最も強い結合が観察され、BA.2、XBB.1.16、および EG.5 S 外部ドメインでは実質的に低いレベルで結合しました (図 6E)。
The differences in the observed trends between the fusion peptide directed antibodies and the stem helix antibody suggest differences in the mechanisms of exposure of their epitopes.
融合ペプチド指向性抗体とステムヘリックス抗体の間で観察された傾向の違いは、それらのエピトープの露出機構の違いを示唆しています。
The substantially stronger binding observed for XBB.1.5 compared to XBB.1.16 or EG.5 further demonstrates the allosteric effect of one or two S1 subunit mutations on the S2 subunit stem-helix epitope.
XBB.1.16またはEG.5と比較してXBB.1.5で観察された実質的に強い結合は、S2サブユニットのステムヘリックスエピトープに対する1つまたは2つのS1サブユニット変異のアロステリック効果をさらに実証しています。
Taken together, our antigenicity data reveal how differently S1 and S2 subunit epitopes have been impacted by SARS-CoV-2 evolution.
まとめると、我々の抗原性データは、S1 サブユニットと S2 サブユニットのエピトープが SARS-CoV-2 の進化によってどのように異なる影響を受けたかを明らかにしています。
Widespread loss in binding was observed to S1-directed antibodies, leaving only selected epitopes such as those on the RBD outer face that bind to antibodies like S309 and DH1193 still capable of binding antibodies elicited against the initial Wu.1 spike.
S1 指向性抗体では広範な結合喪失が観察され、選択されたエピトープのみが残されました。たとえば、S309 や DH1193 などの抗体に結合する RBD 外面上のものは、最初の Wu.1 スパイクに対して誘発された抗体に依然として結合することができます。
S2 subunit epitopes have also been affected in large part due to conformational effects emanating from S1 subunit residue chnages.
S2 サブユニットのエピトープも、S1 サブユニット残基の変更から生じる立体構造効果により、大部分が影響を受けています。
Despite a difference of only two S1 subunit residues between the XBB.1.5 and XBB.1.16 S proteins, the differences in their binding to the fusion peptide-directed and stem helix loop directed antibodies demonstrate allosteric effects of the S1 residue substitution on the S2 subunit.
XBB.1.5 と XBB.1.16 S タンパク質間の S1 サブユニット残基の違いは 2 つだけであるにもかかわらず、融合ペプチド指向性抗体とステムヘリックスループ指向性抗体との結合の違いは、S2 サブユニットに対する S1 残基置換のアロステリック効果を示しています。
For the XBB.1.5 and XBB.1.16 S proteins, these differences in S2 subunit antibody binding were consistent with the differences in the S2 subunit observed in their cryo-EM populations (Figure 3-4).
XBB.1.5 および XBB.1.16 S タンパク質の場合、S2 サブユニットの抗体結合におけるこれらの違いは、クライオ EM 集団で観察された S2 サブユニットの違いと一致していました (図 3-4)。
Similarly, despite a single RBD residue substitution between XBB.1.5 and EG.5 S proteins, the two ectodomains show distinct binding profiles to the fusion peptide-directed and stem helix-directed antibodies.
同様に、XBB.1.5 と EG.5 S タンパク質間の単一 RBD 残基置換にも関わらず、2 つの細胞外ドメインは融合ペプチド指向性抗体とステムヘリックス指向性抗体に対して異なる結合プロファイルを示します。
Vector analysis
ベクトル解析
We next utilized a previously described interprotomer vector-based analysis to examine the Omicron sub-lineage S protein domain arrangements (Figure 7).
次に、以前に説明したプロトマー間ベクターベースの分析を利用して、Omicron 亜系統 S タンパク質ドメインの配置を調べました (図 7)。
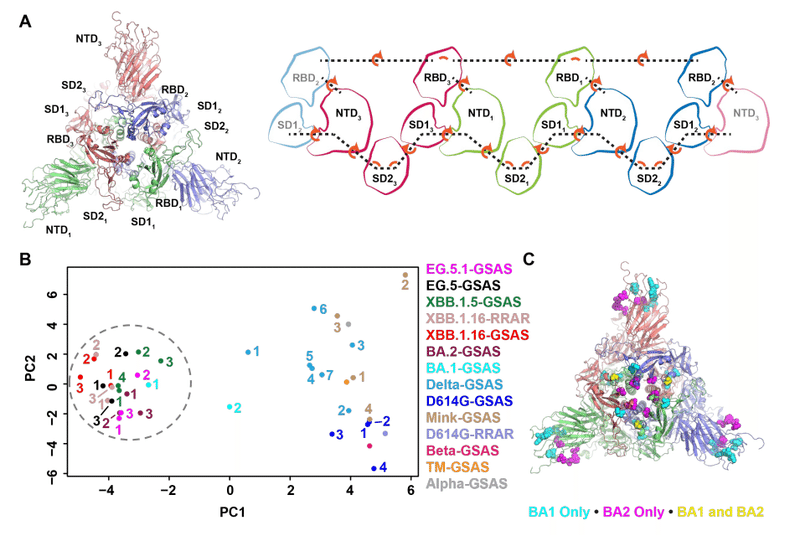
This analysis generates vectors between domains across each chain using domain centroids and anchor residues to define the quaternary geometry of the S protein domains (Figure 7A).
この分析では、ドメイン重心とアンカー残基を使用して各鎖にわたるドメイン間のベクトルを生成し、S タンパク質ドメインの四次構造を定義します (図 7A)。
We showed previously that this geometry differed markedly between MERS, SARS-CoV-1 and SARS-CoV-2 and that these geometries have shifted as SARS-CoV-2 has evolved.
我々は以前、この幾何学形状がMERS、SARS-CoV-1、SARS-CoV-2の間で著しく異なり、SARS-CoV-2が進化するにつれてこれらの幾何学形状が変化したことを示した。
We used these vectors to calculate domain-to-domain distances, angles and dihedrals to determine which vary the most using principal components analysis (PCA).
これらのベクトルを使用してドメイン間の距離、角度、二面角を計算し、主成分分析 (PCA) を使用してどれが最も変化するかを判断しました。
The dataset composed of vectors from the Omicron sub-lineage S ectodomain structures determined here, and our previously determined structures for the D614G, mink, Alpha, Beta, Delta variants and the K417N-E484K-N501Y triple mutant (TM) that appeared in Beta and Gamma VOCs, showed a distinct “Omicron cluster” in the first principal component (Figure 7B).
ここで決定されたオミクロン亜系統の外部ドメイン構造、ベータおよびガンマ VOC に出現する D614G、ミンク、アルファ、ベータ、デルタ変異体および K417N-E484K-N501Y 三重変異体 (TM) の以前に決定された構造からのベクターで構成されるデータセットは、第一主成分に明確な「オミクロンクラスター」を示した (図7B)。
This cluster showed substantially less variability than the earlier variants in both the first and second principal components.
このクラスターは、第 1 主成分と第 2 主成分の両方において、以前のバリアントよりも大幅に低い変動を示しました。
Examination of the vector-based contributions to the first principal component indicated the primary driver of differences between earlier variants and the Omicron variant and sub-lineages were (1) the dihedral orientations between the RBDs, (2) the RBD-to-RBD distances, and (3) the Subdomain 1 (SD1) to S2-central helix distances.
第 1 主成分に対するベクトルベースの寄与を調べると、初期の変異体と Omicron 変異体およびサブ系統の間の差異の主な要因は、(1) RBD 間の二面体の配向、(2) RBD 間の距離、および (3) サブドメイン 1 (SD1) から S2 の中心ヘリックスまでの距離であることが示されました。
A single BA.1 structure (formerly named O1BA.1; PDB: 7TF8) that we previously determined appeared intermediate to the two primary principal component clusters separating the Omicron and earlier variants, while the BA.2 variant structures resided exclusively in the Omicron cluster.
私たちが以前に決定した単一の BA.1 構造 (以前の名前は O1BA.1、PDB: 7TF8) は、Omicron とそれ以前の亜種を分離する 2 つの主主成分クラスターの中間であるように見えました。一方、BA.2 バリアント構造は Omicron クラスターのみに存在しました。
This difference suggests mutations that differ between BA.1 and BA.2 cause the Omicron sub-lineages to settle into a stable domain geometry that was retained in subsequent variants.
この違いは、BA.1 と BA.2 の間で異なる突然変異が、Omicron 亜系統を安定したドメイン形状に定着させ、その後の変異体でも維持されることを示唆しています。
To determine the driving factor in this reduction in domain-to-domain variability in the Omicron sub-lineages, we compared mutation positions between BA.1 and BA.2 that either occurred only in BA.1, only in BA.2, or occurred in both BA.1 and BA.2 but differed in identity (Figure 7C).
Omicron 亜系統におけるドメイン間の変動性の減少の推進要因を特定するために、BA.1 と BA.2 の間で、BA.1 のみ、BA.2 のみ、または BA.1 と BA.2 の両方に発生するが同一性が異なる変異位置を比較しました (図 7C)。
While the BA.1 SD1 T547K mutation may have affected the structure, the BA.2-only RBD-to-RBD interface mutations T376A, D405N, and R408S stood out as likely drivers of the difference between BA.1 and BA.2 that led to the stabilized domain geometry.
BA.1 SD1 T547K 変異は構造に影響を与えた可能性がありますが、BA.2 のみの RBD-to-RBD インターフェース変異 T376A、D405N、および R408S は、安定化されたドメイン ジオメトリをもたらした BA.1 と BA.2 の違いの要因として際立っています。
Taken together, the results from our vector analysis show that close coupling of the RBD domains in the Omicron sub-lineages creates a sterically restricted S1 domain.
まとめると、我々のベクター解析の結果は、Omicron 亜系統における RBD ドメインの緊密な結合により、立体的に制限された S1 ドメインが作成されることを示しています。
Discussion
The emergence of the Omicron variants marked a dramatic inflection point in the COVID-19 pandemic.
Omicron の亜種の出現は、COVID-19パンデミックにおける劇的な転換点を示しました。
The primary characteristic that distinguished Omicron from previous variants was its substantially improved immune evasion.
Omicron を以前の亜種と区別する主な特徴は、免疫回避が大幅に改善されたことでした。
While the pandemic has ended as public health risk has declined, Omicron remains a topic of public health concern as new immune evasive subvariants continue to emerge, necessitating continued surveillance and updated vaccine boosters.
公衆衛生上のリスクが低下したためパンデミックは終息しましたが、新たな免疫回避性亜変異体が出現し続けているため、オミクロンは依然として公衆衛生上の懸念事項であり、継続的な監視と最新のワクチンブースターが必要です。
The most updated booster currently is a monovalent vaccine based on the XBB.1.5 S protein.
現在最も更新されたブースターは、XBB.1.5 S タンパク質に基づく一価ワクチンです。
In this study, we have characterized the structure, receptor binding and antigenicity of the XBB.1.5 S ectodomain, along with the S ectodomains of three XBB lineage members, XBB.1.16, EG.5 and EG.5.1, that followed it.
この研究では、我々は、XBB.1.5 S 外部ドメインとそれに続く 2 つの XBB 系統メンバー、XBB.1.16 EG.5、および EG.5.1 の S 外部ドメインの構造、受容体結合、および抗原性を特徴付けました。
Our data show that the XBB.1.5 RBD is more stable compared to the RBDs of previous variants, as well as the RBD of the EG.5/EG.5.1 sublineage that dominated a subsequent global sweep.
私たちのデータは、XBB.1.5 RBD が以前の亜種の RBD や、その後の世界的大流行を支配した EG.5/EG.5.1 亜系統の RBD と比較してより安定していることを示しています。
A more durable RBD may bode well for the current booster.
より耐久性のある RBD は、現在のブースターにとって良い前兆となる可能性があります。
Additionally, compared to the other Omicron variants tested, the XBB.1.5 S protein showed increased antibody accessibility to subdominant S2 subunit epitopes, particularly at the stem helix region, which may better enable elicitation of potentially protective antibodies that target these regions.
さらに、テストした他の Omicron バリアントと比較して、XBB.1.5 S タンパク質はサブドミナント S2 サブユニット エピトープへの抗体のアクセス可能性が増加したことを示しました。 特にステムヘリックス領域では、これらの領域を標的とする潜在的な防御抗体の誘発がより効果的に可能になる可能性があります。
Our structural studies of the early Omicron S protein variants BA.1 and BA.2 have revealed accumulation of mutations leading to stabilizing interprotomer interactions between down state RBDs, resulting in a tendency to favor the 3-RBD-down conformation that hold the RBD in a closed, receptor inaccessible conformation.
初期の Omicron S タンパク質変異体 BA.1 および BA.2 の構造研究により、ダウン状態の RBD 間のプロトマー間相互作用の安定化につながる変異の蓄積が明らかになりました。 その結果、RBDを閉じた受容体がアクセスできない立体構造に保持する3-RBD-down立体構造を好む傾向が生じます。
This trend, first observed in BA.1, was further fine-tuned in BA.2, where additional mutations optimized the RBD-RBD down state packing, while also stabilizing the RBD itself.
この傾向は、BA.1 で初めて観察され、BA.2 ではさらに微調整され、追加の突然変異により RBD-RBD ダウン状態パッキングが最適化されると同時に、RBD 自体も安定化されました。
This was in stark contrast to previous VOCs with a tendency to favor RBD-up conformations, indicating that among earlier variants transmissibility was a dominant selective force.
これは、RBD-up コンフォメーションを好む傾向があった以前の VOC とはまったく対照的であり、初期の変異体の間では伝達性が支配的な選択力であったことを示しています。
This shift allowed SARS-CoV-2 to remain relevant in an environement where considerable population level immunity had been achieved through vaccination, natural infection or both.
この変化により、ワクチン接種、自然感染、あるいはその両方によってかなりの集団レベルの免疫が達成されていた環境において、SARS-CoV-2が引き続き関連することが可能になった。
The XBB lineage S ectodomains studied here have retained the overall domain organization of the BA.2 S protein, with Omicron S protein structures forming a distinct cluster in our vector analysis that is well-separated from pre-Omicron VOCs (Figure 7).
ここで研究した XBB 系統の外部ドメインは、BA.2 S タンパク質の全体的なドメイン構成を保持しています。Omicron S タンパク質構造は、ベクター分析で明確なクラスターを形成しており、Omicron 以前の VOC から十分に分離されています (図 7)。
The vector analysis highlights the BA.1 S protein architecture to be a transition point with one BA.1 S ectodomain structure residing within the Omicron cluster while another appeared intermediate between the Omicron and pre-Omciron distributions.
ベクター分析により、BA.1 S タンパク質構造が Omicron クラスター内に存在する 1 つの BA.1 S 外部ドメイン構造と、Omicron 分布と Omciron 以前の分布の間の中間に存在するもう 1 つの BA.1 S 外部ドメイン構造による移行点であることが強調されます。
This pattern of progressive transition of the S protein architecture is also demonstrated in Difference Distance Matrix (DDM) analysis, which provides a superposition-free way to compare two structures (Data S5-S6).
S タンパク質構造のこの漸進的遷移パターンは、2 つの構造を重ね合わせずに比較する方法を提供する差分距離行列 (DDM) 解析でも実証されています (データ S5-S6)。
The XBB.1.5, XBB.1.16 and EG.5 S ectodomains are, thus, more similar to the BA.2 S protein, less so to the BA.1 S and least similar to the D614 G S protein.
したがって、XBB.1.5、XBB.1.16、およびEG.5 S 外部ドメインは、BA.2 S タンパク質により類似し、BA.1 S とはそれほど類似せず、D614 GS タンパク質とは最も類似しません。
The RBD “down” conformation hides the most immunodominant and conserved RBD regions, making it an effective immune evasion strategy.
RBD の「ダウン」構造は、最も免疫優勢で保存されている RBD 領域を隠すため、効果的な免疫回避戦略となります。
The stabilization of this 3-RBD-down state, however, presents an apparent conundrum since the RBD-up conformation is rquired for receptor binding and transmission.
しかし、受容体の結合と伝達にはRBD-up立体構造が必要であるため、この3-RBD-down状態の安定化には明らかな難題が存在します。
How does Omicron maintain high affinity receptor binding and transmissibility while retaining and improving the occupancy of its 3-RBD-down state?
Omicron は、3-RBD ダウン状態の占有率を維持および改善しながら、どのようにして高親和性受容体結合と伝達性を維持するのでしょうか?
For the early Omicron BA.1 variant, we had described a strained structure that we envisioned was primed to transition to an open conformation in the presence of receptor.
初期の Omicron BA.1 バリアントについては、受容体の存在下で開いた立体構造に移行する準備が整っていると想定されるひずんだ構造について説明しました。
Our structural conclusions were supported by subsequent HDX-MS studies that revealed a BA.1 spike more readily able to transition to an open state in the presence of the ACE2 receptor.
我々の構造的結論は、BA.1 スパイクが ACE2 受容体の存在下でより容易に開放状態に移行できることを明らかにしたその後の HDX-MS 研究によって裏付けられました。
As Omicron evolved from BA.1 to BA.2, it appeared to have resolved some of the structural strain, although the conformational diversity observed with the XBB.1.5 S 3-RBD-down population, including conformational varibility in the S2 subunit, suggests that internal destabilization or pre-priming may still be a parameter that the evolving Omicron lineage is continuing to optimize.
Omicron が BA.1 から BA.2 に進化するにつれて、構造上の歪みの一部が解決されたように見えましたが、XBB.1.5 S 3-RBD-down 集団で観察された構造の多様性(S2 サブユニットの構造の多様性を含む)は、内部の不安定化またはプレプライミングが依然として進化する Omicron 系統が最適化を続けているパラメーターである可能性があることを示唆しています。
A second factor that enabled Omicron to maintain high affinity receptor binding was the evolution of the RBD itself with mutations within the domain stabilizing the RBD as well as enhancing its intrinsic affinity for ACE2.
Omicron が高親和性の受容体結合を維持できるようにした 2 つ目の要因は、RBD を安定化させるドメイン内の変異による RBD 自体の進化であり、また ACE2 に対する固有の親和性も強化しました。
Finally, a more electropositive host membrane facing surface may have facilitated Omicron S protein interactions with host cell surface coreceptors such as heparan sulfate to increase accumulation of the virus near the host cell and facilitate spike opening.
最後に、より電気陽性の宿主膜に面する表面により、オミクロン S タンパク質とヘパラン硫酸などの宿主細胞表面共受容体との相互作用が促進され、宿主細胞近くのウイルスの蓄積が増加し、スパイクの開口が促進された可能性があります。
An increased electropositive surface on the host membrane facing side was noted for the Omicron BA.1 S protein.
Omicron BA.1 S タンパク質では、宿主膜に面する側の陽性表面の増加が認められました。
For the XBB lineage structures determined here, although there is an overall reduction in positive potential across the surface due to residue subsitutions, the positive electrostatic potential at the center of the host facing surface of the spike is maintained (Figure S9).
ここで決定された XBB 系統構造の場合、残基置換により表面全体で正の電位が全体的に低下しますが、スパイクのホスト側表面の中心の正の静電電位は維持されます (図 S7)。
Indeed, the Omicron variant may be taking a page out of the playbook of the currently endemic coronaviruses that typically adopt the 3-RBD-down conformation with interactions with the host glycocalyx facilitating the transition of the RBD to the up position, thus enabling optimal immune evasion while ensuring the required transition to the conformation that can bind host receptor.
実際、Omicron 変異体は、一般的に 3-RBD-down 構造をとり、宿主の糖衣との相互作用によって RBD の上位への移行を促進する、現在流行しているコロナウイルスの戦略の 1 ページを取り上げている可能性があります。 したがって、宿主受容体に結合できる立体構造への必要な移行を確実にしながら、最適な免疫回避が可能になります。
The ongoing evolution of the Omicron variant spikes involves multi-parameter optimization where the major driver is immune evasion.
Omicron バリアント スパイクの継続的な進化には、主な要因が免疫回避であるマルチパラメータの最適化が含まれています。
The other properties that need to be simultaneously optimized include pre-fusion conformation stability, host receptor binding and ability to undergo the conformation changes required for entry into host cell.
同時に最適化する必要がある他の特性には、融合前の立体構造安定性、宿主受容体結合、宿主細胞への侵入に必要な立体構造変化を受ける能力などがあります。
Our study shows how the XBB lineage gained increased immune evasive properties while improving spike stability and receptor binding, and how it has continued to evolve.
私たちの研究は、XBB 系統がスパイクの安定性と受容体結合を改善しながら免疫回避特性をどのように獲得したか、そしてそれがどのように進化し続けてきたかを示しています。
The substantial structural, conformational and antigenic impact that were effected by only one or two residue substitutions underscored the effectiveness of allosteric networks in the SARS-CoV-2 S protein and foretell its continued evolution to escape host immunity.
わずか 1 つまたは 2 つの残基置換によって影響を受ける実質的な構造的、立体構造的、および抗原性の影響は、SARS-CoV-2 S タンパク質におけるアロステリック ネットワークの有効性を強調し、宿主免疫から逃れるためのその継続的な進化を予告しています。
以下省略。
この記事が気に入ったらサポートをしてみませんか?