JN.1、KP.2、KP.3の変異プロファイリングと結合自由エネルギー分析により、ACE2 親和性と免疫回避のエピスタシス ホットスポットが明らかに

Atomistic Prediction of Structures, Conformational Ensembles and Binding Energetics for the SARS-CoV-2 Spike JN.1, KP.2 and KP.3 Variants Using AlphaFold2 and Molecular Dynamics Simulations: Mutational Profiling and Binding Free Energy Analysis Reveal Epistatic Hotspots of the ACE2 Affinity and Immune Escape
SARS-CoV-2 スパイク JN.1、KP.2、KP.3 変異体の構造、立体配座アンサンブル、結合エネルギーの原子レベルでの予測 (AlphaFold2 と分子動力学シミュレーションを使用):変異プロファイリングと結合自由エネルギー分析により、ACE2 親和性と免疫回避のエピスタシス ホットスポットが明らかになる。
Abstract
The most recent wave of SARS-CoV-2 Omicron variants descending from BA.2 and BA.2.86 exhibited improved viral growth and fitness due to convergent evolution of functional hotspots.
BA.2 および BA.2.86 から派生した SARS-CoV-2 オミクロン変異体の最新の波は、機能的ホットスポットの収斂進化により、ウイルスの成長と適応度が向上しました。
These hotspots operate in tandem to optimize both receptor binding for effective infection and immune evasion efficiency, thereby maintaining overall viral fitness.
これらのホットスポットは連携して機能し、効果的な感染と免疫回避効率の両方の受容体結合を最適化し、それによって全体的なウイルスの適応度を維持します。
The lack of molecular details on structure, dynamics and binding energetics of the latest FLiRT and FLuQE variants with the ACE2 receptor and antibodies provides a considerable challenge that is explored in this study.
最新の FLiRT および FLuQE 変異体と ACE2 受容体および抗体の構造、ダイナミクス、結合エネルギーに関する分子の詳細が不足しているため、この研究ではかなりの課題が検討されています。
We combined AlphaFold2-based atomistic predictions of structures and conformational ensembles of the SARS-CoV-2 Spike complexes with the host receptor ACE2 for the most dominant Omicron variants JN.1, KP.1, KP.2 and KP.3 to examine the mechanisms underlying the role of convergent evolution hotspots in balancing ACE2 binding and antibody evasion.
我々は、最も優勢なオミクロン変異体 JN.1、KP.1、KP.2、KP.3 について、SARS-CoV-2 スパイク複合体の構造とコンフォメーション アンサンブルの AlphaFold2 ベースの原子予測と宿主受容体 ACE2 を組み合わせ、ACE2 結合と抗体回避のバランスをとる収束進化ホットスポットの役割の根底にあるメカニズムを調べました。
Using the ensemble-based mutational scanning of the spike protein residues and computations of binding affinities, we identified binding energy hotspots and characterized molecular basis underlying epistatic couplings between convergent mutational hotspots.
スパイク タンパク質残基のアンサンブル ベースの変異スキャンと結合親和性の計算を使用して、結合エネルギー ホットスポットを特定し、収束変異ホットスポット間のエピスタシス結合の根底にある分子基盤を特徴付けました。
The results suggested that the existence of epistatic interactions between convergent mutational sites at L455, F456, Q493 positions that enable to protect and restore ACE2 binding affinity while conferring beneficial immune escape.
結果は、L455、F456、Q493の位置にある収束変異部位間のエピスタシス相互作用の存在が、ACE2結合親和性を保護および回復しながら有益な免疫回避をもたらすことを示唆した。
To examine immune escape mechanisms, we performed structure-based mutational profiling of the spike protein binding with several classes of antibodies that displayed impaired neutralization against BA.2.86, JN.1, KP.2 and KP.3.
免疫回避メカニズムを調べるために、BA.2.86、JN.1、KP.2、KP.3 に対する中和が損なわれたいくつかのクラスの抗体とスパイクタンパク質の結合の構造ベースの変異プロファイリングを実行した。
The results confirmed the experimental data that JN.1, KP.2 and KP.3 harboring the L455S and F456L mutations can significantly impair the neutralizing activity of class-1 monoclonal antibodies, while the epistatic effects mediated by F456L can facilitate the subsequent convergence of Q493E changes to rescue ACE2 binding.
結果は、L455SおよびF456L変異を有するJN.1、KP.2、およびKP.3がクラス1モノクローナル抗体の中和活性を著しく損なう可能性がある一方で、F456Lによって媒介されるエピスタシス効果は、その後のQ493E変化の収束を促進してACE2結合を回復できるという実験データを裏付けました。
Structural and energetic analysis provided a rationale to the experimental results showing that BD55-5840 and BD55-5514 antibodies that bind to different binding epitopes can retain neutralizing efficacy against all examined variants BA.2.86, JN.1, KP.2 and KP.3.
構造およびエネルギー分析により、異なる結合エピトープに結合するBD55-5840およびBD55-5514抗体が、検査されたすべての変異体BA.2.86、JN.1、KP.2、およびKP.3に対して中和効果を維持できることを示した実験結果の根拠が示されました。
The results support the notion that evolution of Omicron variants may favor emergence of lineages with beneficial combinations of mutations involving mediators of epistatic couplings that control balance of high ACE2 affinity and immune evasion.
結果は、オミクロン変異体の進化が、高いACE2親和性と免疫回避のバランスを制御するエピスタシス結合のメディエーターを含む変異の有益な組み合わせを持つ系統の出現を促進する可能性があるという考えを支持しています。
1. Introduction
The Spike (S) glycoprotein of SARS-CoV-2 plays a pivotal role in the virus's ability to enter host cells.
SARS-CoV-2 のスパイク (S) 糖タンパク質は、ウイルスが宿主細胞に侵入する能力において極めて重要な役割を果たします。
The wealth of structural and biochemical investigations conducted on the S glycoprotein has provided crucial insights into the mechanisms that regulate virus transmission and immune evasion.
S 糖タンパク質について実施された豊富な構造的および生化学的調査は、ウイルスの伝染と免疫回避を制御するメカニズムに関する重要な洞察を提供しました。
The conformational flexibility of the S glycoprotein, particularly within the S1 subunit and its various domains , which includes the N-terminal domain (NTD), the receptor-binding domain (RBD), and two structurally conserved subdomains—SD1 and SD2, allows it to adapt to different stages of the viral entry process.
S 糖タンパク質の構造的柔軟性、特に S1 サブユニットとそのさまざまなドメイン (N 末端ドメイン (NTD)、受容体結合ドメイン (RBD)、および構造的に保存された 2 つのサブドメイン (SD1 と SD2) を含む) により、ウイルス侵入プロセスのさまざまな段階に適応できます。
The transitions between closed and open states, driven by conformational changes in the NTD and RBD, enable the virus to effectively engage with host cell receptors while evading immune surveillance through structural variability.
NTDとRBDの構造変化によって引き起こされる閉鎖状態と開放状態の間の遷移により、ウイルスは構造的変動性を通じて免疫監視を回避しながら宿主細胞受容体と効果的に結合することができます。
Understanding these structural dynamics is crucial for designing therapeutics and vaccines that target the S protein, aiming to disrupt viral entry and prevent infection by SARS-CoV-2.
これらの構造ダイナミクスを理解することは、ウイルスの侵入を阻止し、SARS-CoV-2 による感染を防ぐことを目的とした、S タンパク質を標的とした治療薬やワクチンを設計する上で非常に重要です。
Biophysical investigations have delineated how thermodynamic principles and kinetic factors govern the mechanisms of the S protein.
生物物理学的調査により、熱力学的原理と運動学的因子がSタンパク質のメカニズムをどのように制御しているかが明らかにされている。
These studies have revealed that mutations within the S protein, particularly in the S1 subunit, can induce structural alterations that affect its stability and conformational dynamics, particularly the protein's ability to switch between the open and closed states that can influence the accessibility of the RBD critical for viral attachment to host cells.
これらの研究により、Sタンパク質、特にS1サブユニット内の変異は、その安定性と立体構造のダイナミクス、特にウイルスが宿主細胞に付着するために重要なRBDへのアクセス性に影響を与える可能性のある、タンパク質の開状態と閉状態を切り替える能力に影響を与える構造変化を引き起こす可能性があることが明らかになりました。
Moreover, long-range interactions between the dynamic S1 subunit (including domains like the N-terminal domain and RBD) and the more rigid S2 subunit (involved in membrane fusion) play crucial roles in determining the overall architecture and functional states of the S protein trimer.
さらに、動的なS1サブユニット(N末端ドメインやRBDなどのドメインを含む)とより剛性の高いS2サブユニット(膜融合に関与)との間の長距離相互作用は、Sタンパク質三量体の全体的な構造と機能状態を決定する上で重要な役割を果たします。
The emergence and evolution of SARS-CoV-2 variants like BQ.1.1 and XBB.1 have raised significant interest due to their distinct characteristics, including superior growth advantages and potential immune evasion capabilities.
BQ.1.1やXBB.1のようなSARS-CoV-2変異体の出現と進化は、優れた成長上の利点や潜在的な免疫回避能力など、その独特の特徴により大きな関心を集めています。
The XBB.1.5 subvariant has evolved through recombination events within the BA.2 lineage, incorporating genetic material from BA.2.10.1 and BA.2.75 sublineages.
XBB.1.5 亜変異体は、BA.2 系統内での組み換えイベントを通じて進化し、BA.2.10.1 および BA.2.75 亜系統の遺伝物質を組み込んでいます。
XBB.1.5 is equally immune evasive as XBB.1 but may have growth advantage by virtue of the higher ACE2 binding as F486P in the XBB.1.5 subvariant can restore most of the favorable hydrophobic contacts.
XBB.1.5 は XBB.1 と同様に免疫回避性がありますが、ACE2 結合性が高いため、成長に有利である可能性があります。 XBB.1.5 亜変異体の F486P は、好ましい疎水性接触のほとんどを回復できるためです。
Further investigations have substantiated that the enhanced growth and increased transmissibility observed in the XBB.1.5 lineage likely stem from its preserved resistance to neutralization and improved affinity for binding to ACE2 receptors.
さらなる調査により、XBB.1.5系統で観察された増殖の促進と伝染性の増加は、中和に対する耐性が維持され、ACE2受容体への結合親和性が向上したことに起因する可能性が高いことが実証されています。
By October 2023, XBB sublineages such as XBB.1.5 and XBB.1.16, both featuring the F486P substitution, had become prevalent globally (source: https://nextstrain.org/).
2023年10月までに、F486P置換を特徴とするXBB.1.5やXBB.1.16などのXBB亜系統が世界中で普及しました(出典:https://nextstrain.org/)。
Compared to XBB.1.5, XBB.1.16 exhibits two substitutions: E180V in the NTD and T478R in the receptor-RBD.
XBB.1.5と比較すると、XBB.1.16はNTDのE180Vと受容体RBDのT478Rの2つの置換を示しています。
These emerging variants demonstrated increased infectivity and transmissibility compared to earlier Omicron variants.
これらの新たな変異体は、以前のオミクロン変異体と比較して感染性と伝染性が向上していることが実証されています。
Moreover, several residues in the RBD (R346, K356, K444, V445, G446, N450, L452, N460, F486, F490, R493, and S494) are mutated in at least five distinct new Omicron lineages.
さらに、RBD のいくつかの残基 (R346、K356、K444、V445、G446、N450、L452、N460、F486、F490、R493、および S494) は、少なくとも 5 つの異なる新しいオミクロン系統で変異しています。
The lineage of XBB descendants, including EG.5 and EG.5.1, which carry an additional mutation F456L, has become one of the dominant lineages currently.
EG.5 および EG.5.1 を含む XBB 子孫の系統は、追加の変異 F456L を持ち、現在、優勢な系統の 1 つになっています。
EG.5 evolved from Omicron XBB.1.9 and bears only one additional substitution, F456L, compared to XBB.1.5.
EG.5 はオミクロン XBB.1.9 から進化し、XBB.1.5 と比較して、1 つの追加置換 F456L のみを持ちます。
Its direct descendant, EG.5.1, features Q52H in the NTD and F456L in the RBD.
その直接の子孫である EG.5.1 は、NTD に Q52H、RBD に F456L を備えています。
EG.5 and EG.5.1 were discovered to exhibit moderate resistance to antibody neutralization compared to XBB.1.5 and this resistance is particularly pronounced against class 1 monoclonal antibodies, primarily due to a single F456L mutation in the RBD.
EG.5 と EG.5.1 は、XBB.1.5 と比較して抗体中和に対して中程度の耐性を示すことが発見され、この耐性はクラス 1 モノクローナル抗体に対して特に顕著です。 主に RBD の単一の F456L 変異が原因です。
Further studies specifically on the immune evasion of the EG.5.1 subvariant confirmed that the enhanced neutralization escape observed is primarily driven by the F456L mutation rather than the Q52H mutation.
EG.5.1 亜変異体の免疫回避に関するさらなる研究により、観察された中和回避の強化は、Q52H 変異ではなく F456L 変異によって主に引き起こされていることが確認されました。
XBB subvariants carrying both the L455F and F456L combination of flipped substitutions are often referred to as "FLip" variants and these variants were identified in over 20% of global XBB samples collected at the beginning of September 2023.
L455FとF456Lの反転置換の組み合わせを持つXBBサブバリアントは、しばしば「FLip」バリアントと呼ばれ、これらのバリアントは、2023年9月初旬に収集された世界中のXBBサンプルの20%以上で特定されました。
The FLip variants include JG.3 (XBB.1.9.2.5.1.3.3), JF.1 (XBB.1.16.6.1), GK.3 (XBB.1.5.70.3), and JD.1.1, all of which emerged convergently.
FLip 変異体には、JG.3 (XBB.1.9.2.5.1.3.3)、JF.1 (XBB.1.16.6.1)、GK.3 (XBB.1.5.70.3)、JD.1.1 があり、これらはすべて収束的に出現しました。
This convergence underscores that acquiring the L455F/F456L double mutation can provide a growth advantage to XBB within the human population.
この収束は、L455F/F456L 二重変異を獲得すると、ヒト集団内で XBB の成長に有利になる可能性があることを強調している。
The Omicron subvariant BA.2.86, originating from the BA.2 variant, shows substantial genetic divergence from earlier forms (Table1, Figure 1).
BA.2 変異体から派生したオミクロン亜変異体 BA.2.86 は、以前の形態とは遺伝的に大きく異なっています (表 1、図 1)。
Biophysical investigations have confirmed that the BA.2.86 variant can resist neutralization by monoclonal antibodies targeting epitopes in the NTD, SD1, as well as classes 1, 2, and 3 epitopes within the RBD.
生物物理学的調査により、BA.2.86 変異体は、NTD、SD1 のエピトープ、および RBD 内のクラス 1、2、3 エピトープを標的とするモノクローナル抗体による中和に抵抗できることが確認されました。
Notably, BA.2.86 exhibits a greater potential to evade RBD-targeted antibodies compared to the immune evasion observed in XBB.1.5 and EG.5.1 variants.
注目すべきことに、BA.2.86は、XBB.1.5およびEG.5.1変異体で観察された免疫回避と比較して、RBD標的抗体を回避する可能性が高くなっています。
The immune evasion capability of the BA.2.86 subvariant was evaluated using a panel of neutralizing antibodies that are effective against XBB.1.5, revealing that BA.2.86 can evade antibodies that target XBB.1.5.
BA.2.86 亜変異体の免疫回避能力は、XBB.1.5 に有効な中和抗体パネルを使用して評価され、BA.2.86 は XBB.1.5 を標的とする抗体を回避できることが明らかになりました。
Recent studies investigating the structure and binding characteristics of the BA.2.86 spike protein with ACE2 and antibodies have shown that the mutations acquired by BA.2.86 do not result in significant changes in antibody evasion compared to XBB.1.5.
BA.2.86スパイクタンパク質とACE2および抗体の構造および結合特性を調査した最近の研究では、BA.2.86が獲得した変異は、XBB.1.5と比較して抗体回避に大きな変化をもたらさないことが示されています。
However, the RBD of BA.2.86 displays a 2.2-fold increase in affinity for ACE2 compared to XBB.1.5, thereby providing BA.2.86 with a transmission advantage.
しかし、BA.2.86のRBDは、XBB.1.5と比較してACE2に対する親和性が2.2倍に増加しており、BA.2.86に伝播上の利点を与えている。
The latest cryo-EM structure of ACE2 complexed with BA.2.86 trimeric S protein supported the notion that the enhanced binding affinity of BA.2.86 may be driven by the electrostatic complementarity between BA.2.86 RBD and ACE2 and also potentially enhanced by the flexibility of the RBDs, allowing better exposure of the ACE2 binding within the trimer structure.
BA.2.86三量体Sタンパク質と複合体を形成したACE2の最新のクライオ電子顕微鏡構造は、BA.2.86の結合親和性の向上は、BA.2.86 RBDとACE2間の静電的相補性によってもたらされ、RBDの柔軟性によっても強化され、三量体構造内でACE2結合がよりよく露出される可能性があるという考えを裏付けた。
Another study also suggested that BA.2.86 does not have greater immune escape relative t XBB.1.5 from neutralizing immunity elicited by either Omicron XBB-family subvariant infection but improves virus fitness through greater binding affinity to ACE2.
別の研究でも、BA.2.86 は、Omicron XBB ファミリー亜種感染のいずれかによって誘発される中和免疫からの免疫回避能力が XBB.1.5 に比べて高くないが、ACE2 への結合親和性が高いためウイルスの適応度が向上することが示唆されている。
JN.1 is a variant of BA.2.86 which emerged independently from Omicron BA.2 and harbors an additional L455S mutation responsible for enhanced immune escape.
JN.1 は BA.2.86 の変異体で、Omicron BA.2 とは独立して出現し、免疫回避の強化に関与する追加の L455S 変異を抱えています。
BA.2.86/JN.1 also showed a long genetic distance from XBB.1.5 and XBB.1.16 (Table 1, Figure 1).
BA.2.86/JN.1 は、XBB.1.5 および XBB.1.16 と遺伝的に遠いことも示しました (表 1、図 1)。
The JN.1 variant is antigenically distinct from the XBB.1.5 variant.
JN.1 変異体は、XBB.1.5 変異体とは抗原的に異なります。
A comparative biochemical analysis using surface plasmon resonance (SPR) assays showed a notable reduction in ACE2 binding affinity for JN.1 indicating that its enhanced immune evasion capabilities come at the expense of reduced ACE2 binding.
表面プラズモン共鳴 (SPR) アッセイを使用した比較生化学分析では、JN.1 に対する ACE2 結合親和性が著しく低下していることが示され、免疫回避能力の強化は ACE2 結合の低下を犠牲にしていることが示されました。
Despite only a single additional mutation (L455S) compared to its predecessor BA.2.86, which results in increased resistance to humoral immunity, the JN.1 variant swiftly became predominant in Europe and outcompeted the previously dominant XBB lineage by early 2024.
JN.1変異体は、その前身であるBA.2.86と比較して、体液性免疫に対する抵抗性を高める1つの追加変異(L455S)があるだけであるにもかかわらず、ヨーロッパで急速に優勢となり、2024年初頭までにそれまで優勢であったXBB系統に打ち勝ちました。
In vitro ACE2 binding assay showed that the dissociation constant value of the JN.1 RBD was significantly higher than that of the BA.2.86 RBD indicating that L455S mutation leads to the decreased binding affinity while it displays robust immune resistance, particularly against antibodies induced by the XBB.1.5 vaccine.
インビトロACE2結合アッセイでは、JN.1 RBDの解離定数値がBA.2.86 RBDよりも有意に高いことが示され、L455S変異により結合親和性が低下する一方で、特にXBB.1.5ワクチンによって誘導される抗体に対して強力な免疫抵抗性を示すことが示された。
JN.1 demonstrated increased evasion against RBD class-1 antibodies, such as S2K146 and Omi-18, and the class-3 antibody S309 corroborating findings from other related studies.
JN.1 は、S2K146 や Omi-18 などの RBD クラス 1 抗体、およびクラス 3 抗体 S309 に対する回避能力が高まっていることが示され、他の関連研究の知見を裏付けています。
A series of variants with mutations at L455, F456, and R346 convergent hotspot sites emerged including “SLip” variant which has the JN.1 mutations (L455S) with the additional F456L mutation.
L455、F456、R346の収束ホットスポット部位に変異を持つ一連の変異体が出現し、その中にはJN.1変異(L455S)に加えてF456L変異も持つ「SLip」変異体も含まれている。
More recently, we have seen the emergence of the FLiRT variant, which harbors an additional R346T mutation in the backbone of SLip.
さらに最近では、SLipのバックボーンにR346T変異をさらに持つFLiRT変異体の出現がみられました。
Recent studies revealed that that F456L (SLip) and R346T (FLiRT) subvariants of JN.1 contribute to further escape of JN.1- derived variants from neutralizing antibodies.
最近の研究では、JN.1のF456L(SLip)およびR346T(FLiRT)サブバリアントが、JN.1由来の変異体が中和抗体からさらに逃れるのに寄与していることが明らかになりました。
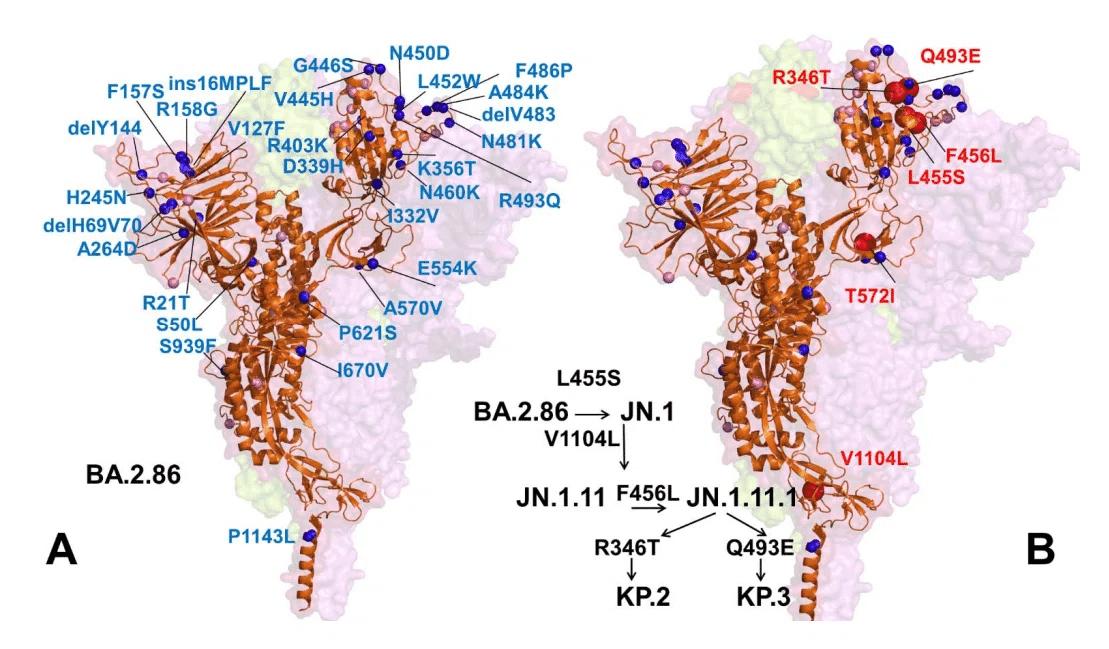
Since January 2024, JN.1 diversified into a number of sublineages, many of which share recurrent mutations R346T ( JN.1.18), F456L ( JN.1.16), T572I ( JN.1.7), or combinations of these mutations (KP.2 variant).
2024年1月以降、JN.1はいくつかの亜系統に多様化し、その多くはR346T(JN.1.18)、F456L(JN.1.16)、T572I(JN.1.7)、またはこれらの変異の組み合わせ(KP.2変異体)を共有しています。
JN.1 subvariants including KP.2 (JN.1.11.1.2) and KP.3 (JN.1.11.1.3), which convergently acquired S protein substitutions such as S:R346T, S:F456L, and S:Q493E, and V1104L have emerged concurrently.
S:R346T、S:F456L、S:Q493E、V1104LなどのSタンパク質置換を収束的に獲得したKP.2(JN.1.11.1.2)およびKP.3(JN.1.11.1.3)を含むJN.1亜種が同時に出現しました。
KP.2 and KP.3 are members of a “FLiRT” group of variants.
KP.2 と KP.3 は「FLiRT」変異体グループのメンバーです。
Other FLiRT variants, including KP.1.1, have also been identified as circulating in the U.S., but have not yet become as widespread as KP.2 or KP.3.
KP.1.1 を含む他の FLiRT 変異体も米国で流行していることが確認されていますが、KP.2 や KP.3 ほど広まってはいません。
JN.1 subvariants with one or more of these recurrent mutations, such as KP.2 (R346T, F456L and V1104L), appear to have a growth advantage, becoming more dominant in multiple regions as of April 2024.
KP.2(R346T、F456L、V1104L)など、これらの反復変異を1つ以上持つJN.1亜種は、成長上の利点があり、2024年4月現在、複数の地域でより優勢になっているようです。
Virological properties of KP.2 variant were examined showing that that the infectivity of KP.2 is significantly lower than that of JN.1 and that KP.2 has an increased immune resistance ability compared to JN.1.
KP.2 変異体のウイルス学的特性を調べたところ、KP.2 の感染性は JN.1 よりも大幅に低く、KP.2 は JN.1 に比べて免疫抵抗能力が高まっていることがわかった。
Another JN.1 descendant termed "FLuQE" variants (KP.3) continues to dominate "FLiRT" and show strong growth. KP.3 (JN.1.11.1.3), featured mutations R346T, L455S, F456L, Q493E and V1104L.
JN.1の別の子孫である「FLuQE」変異体(KP.3)は、「FLiRT」を支配し続けており、力強い成長を示しています。KP.3(JN.1.11.1.3)は、R346T、L455S、F456L、Q493E、およびV1104Lの変異を特徴としています。
The pseudovirus infectivity of KP.2 and KP.3 was significantly lower than that of JN.1.
KP.2およびKP.3の擬似ウイルスの感染性は、JN.1の感染性よりも大幅に低かった。
Furthermore, JN.1 subvariants such as LB.1 and KP.2.3, which convergently acquired S31 deletion in addition to the above substitutions, have emerged and spread as of June 2024 and contribute to immune evasion and the increased relative effective reproduction number.
さらに、上記の置換に加えてS31欠失を収束的に獲得したLB.1やKP.2.3などのJN.1亜種が2024年6月現在出現し、拡散しており、免疫回避と相対有効再生産数の増加に寄与している。
Importantly, LB.1 and KP.2.3 exhibited higher pseudovirus infectivity and more robust immune resistance than KP.2 showing that S31del is important for the increased infectivity and enhanced immune evasion.
重要なのは、LB.1 と KP.2.3 は KP.2 よりも高い擬似ウイルス感染性とより強力な免疫抵抗性を示したことで、S31del が感染性の増加と免疫回避の強化に重要であることを示したことです。
Both KP.2 and KP.3 variants share the F456L mutation that is critical for enhancing antibody evasion by impairing binding to class 1 RBD antibodies, confirming that antibody evasion can represent a major selective advantage for viral spread.
KP.2 と KP.3 の両方の変異体は、クラス 1 RBD 抗体への結合を阻害することで抗体回避を強化するために重要な F456L 変異を共有しており、抗体回避がウイルス拡散の大きな選択的利点になり得ることを確認しています。
According to recent findings from Cao lab and private communications of June 4 (https://twitter.com/yunlong_cao) KP.3 variant is starting to outcompete KP.2 due to Q493E mutation that enables the higher ACE2 binding affinity than KP.2 and enhanced immune evasion primarily against class I of RBD antibodies.
Cao 研究室の最近の調査結果と 6 月 4 日の非公開の通信 (https://twitter.com/yunlong_cao) によると、KP.3 変異体は、KP.2 よりも高い ACE2 結合親和性と、主に RBD 抗体のクラス I に対する免疫回避の強化を可能にする Q493E 変異により、KP.2 に打ち勝ち始めています。
Moreover, KP.3 (JN.1+F456L+Q493E) is the most immune evasive variant and is also the fastest-growing JN.1 sublineage.
さらに、KP.3 (JN.1+F456L+Q493E) は最も免疫回避性の高い変異体であり、最も急速に成長している JN.1 サブ系統でもあります。
The additional F456L and Q493E mutation allows KP.3 to evade a substantial proportion of JN.1-effective antibodies, especially Class 1 antibodies.
追加の F456L および Q493E 変異により、KP.3 は JN.1 に有効な抗体、特にクラス 1 抗体のかなりの割合を回避できます。
Deep mutational scanning (DMS) experiments and functional studies suggested that evolutionary windows for the Omicron variants could be enhanced through epistatic interactions between variant mutations where the effect of one mutation can be altered depending on the presence of other mutations, resulting in non-additive impacts of mutations on specific functions.
ディープミューテーションスキャニング(DMS)実験と機能研究により、オミクロン変異体の進化の窓は、変異体変異間のエピスタシス相互作用を通じて強化される可能性があることが示唆されました。この相互作用では、1つの変異の影響が他の変異の存在に応じて変化する可能性があり、その結果、特定の機能に対する変異の非加法的影響が生じます。
These experiments showed evidence of compensatory epistasis in which immune escape mutations can individually reduce ACE2 binding but are compensated through epistatic couplings with affinity-enhancing Q498R and N501Y mutations.
これらの実験は、免疫逃避変異が個別にACE2結合を低下させる可能性があるが、親和性を高めるQ498RおよびN501Y変異とのエピスタシス結合によって補償されるという補償的エピスタシスの証拠を示した。
Recent DMS experiments examined the impacts of all mutational changes and single-codon deletions in the XBB.1.5 RBDs on ACE2-binding affinity and RBD folding efficiency, revealing the expanded character of epistatic couplings between RBD residues including epistatic interactions between R493Q reversed mutations and mutations at positions Y453, L455, and F456.
最近の DMS 実験では、XBB.1.5 RBD のすべての変異変化と単一コドン欠失が ACE2 結合親和性と RBD 折り畳み効率に与える影響が調べられました。 R493Q 逆転変異と位置 Y453、L455、および F456 の変異間のエピスタシス相互作用を含む、RBD 残基間のエピスタシス結合の拡張された特性が明らかになりました。
It was also demonstrated through mutational surveys of Omicron variants up until BA.2.86 that Q493E is typically detrimental to ACE2 binding, often incurring up to 10-fold of loss in the binding affinity.
また、BA.2.86までのオミクロン変異体の変異調査を通じて、Q493Eは典型的にはACE2結合に有害であり、結合親和性が最大10倍低下することが多いことが実証されました。
In recent correspondence, Starr and colleagues reported that when Q493E mutation is combined with L455S and F456L mutations of FliRT variants, the loss of binding affinity is reversed and instead showed the increased ACE2 binding for KP.3 (https://x.com/tylernstarr/status/1800315116929560965).
最近の通信で、Starr と同僚は、Q493E 変異を FliRT 変異体の L455S および F456L 変異と組み合わせると、結合親和性の喪失が逆転し、代わりに KP.3 に対する ACE2 結合が増加することを報告しました。
Moreover, Starr and colleagues reported on the results of human ACE2 binding assays on all combinations of L455S, F456L, and Q493E mutations in the BA.2.86 background, revealing the reversal of the deleterious effect of Q493E in the background of L455S+F456L
さらに、Starr 氏と同僚は、BA.2.86 バックグラウンドにおける L455S、F456L、Q493E 変異のすべての組み合わせに対するヒト ACE2 結合アッセイの結果を報告し、L455S+F456L バックグラウンドにおける Q493E の有害な影響が逆転したことを明らかにしました。
The most recent reports from Cao’s lab showed that enhanced receptor ACE2 binding capabilities of the KP.3 variant may be enabled by a substantial synergistic effect of F456L and Q493E mutations.
Cao 氏の研究室からの最新の報告では、KP.3 変異体の受容体 ACE2 結合能力の向上は、F456L 変異と Q493E 変異の大きな相乗効果によって可能になる可能性があることが示されました。
While F456L (KD = 12 nM) and R346T + F456L (KD = 11 nM) did not largely affect the hACE2-binding affinity of JN.1 (KD = 13 nM), Q493E mutation of KP.3 substantially improved the receptor binding affinity showing KD = 6.9 nM which indicates non-additive epistatic interactions between Q493E and other mutations of KP.3, particularly F456L in comparison with these pre-BA.2.86 variants.
F456L(KD = 12 nM)およびR346T + F456L(KD = 11 nM)はJN.1(KD = 13 nM)のhACE2結合親和性に大きな影響を与えなかったが、KP.3のQ493E変異は受容体結合親和性を大幅に改善し、KD = 6.9 nMを示した。これは、これらのpre-BA.2.86変異体と比較して、Q493EとKP.3の他の変異、特にF456Lとの間に非相加的エピスタシス相互作用があることを示す。
This insightful study also shows that high affinity of KP.3 due epistasis, may enable the incorporation of A475V ( this mutation is convergently observed in JD.1.1, HK.3.14, JN.4, and KP.2.3.1 subvariants) for further immune evasion, given the observed only small reduction in ACE2 binding affinity for KP.3 + A475V (KD is 22 nM).
この洞察に満ちた研究は、エピスタシスによるKP.3の高い親和性により、KP.3 + A475Vに対するACE2結合親和性のわずかな低下(KDは22 nM)が観察されたことを考慮すると、さらなる免疫回避のためにA475V(この変異はJD.1.1、HK.3.14、JN.4、およびKP.2.3.1サブバリアントで収束的に観察される)が組み込まれる可能性があることも示している。
This study indicated that the high ACE2-binding affinity of KP.3 variant may drive the rapid transmission and prevalence of this subvariant and its descendants, enhancing its potential to acquire additional immune-evasive mutations.
この研究は、KP.3変異体のACE2結合親和性の高さが、この亜種とその子孫の急速な伝播と蔓延を促進し、免疫回避変異をさらに獲得する可能性を高めている可能性を示唆した。
Several other recent studies confirmed that while JN.1 displays lower affinity to ACE2 and higher immune evasion properties compared to BA.2.86, indicating that new emerging subvariants bearing Q493E mutation in the background of FLiRT variants could recover ACE2 binding affinity and continue to display the enhanced antibody evasion profile.
最近のいくつかの研究では、JN.1はBA.2.86と比較してACE2への親和性が低く、免疫回避特性が高いことが確認されており、FLiRT変異体を背景にQ493E変異を持つ新たな亜変異体はACE2結合親和性を回復し、強化された抗体回避プロファイルを示し続ける可能性があることが示唆されています。
Computer simulations provided important atomistic and mechanistic advances into understanding the dynamics and function of the SARS-CoV-2 S proteins.
コンピューターシミュレーションは、SARS-CoV-2 Sタンパク質のダイナミクスと機能を理解する上で重要な原子論的および機構的進歩をもたらしました。
A series of experimental and computational studies revealed that the SARS-CoV-2 S protein can function as an allosteric regulatory machinery that is controlled by stable allosteric hotspots to modulate specific regulatory and binding functions.
一連の実験的および計算的研究により、SARS-CoV-2 Sタンパク質は、安定したアロステリックホットスポットによって制御され、特定の調節機能と結合機能を調節するアロステリック調節機構として機能できることが明らかになりました。
Our recent studies demonstrated that convergent Omicron mutations such as G446S, F486V, F486P, F486S, and F490S can display epistatic couplings with the major stability and binding affinity hotspots which may allow for the observed broad antibody resistance Analysis of conformational dynamics, binding and allosteric communications in the Omicron S protein complexes with the ACE2 host receptor characterized regions of epistatic couplings that are centered at the binding affinity hotspots N501Y and Q498R.
我々の最近の研究では、G446S、F486V、F486P、F486S、F490Sなどの収束性オミクロン変異は、主要な安定性および結合親和性のホットスポットとのエピスタシス結合を示す可能性があり、観察された広範な抗体耐性を可能にする可能性があることが実証されました。ACE2宿主受容体とのオミクロンSタンパク質複合体におけるコンフォメーションダイナミクス、結合、およびアロステリックコミュニケーションの分析は、結合親和性ホットスポットN501YおよびQ498Rを中心とするエピスタシス結合の領域を特徴付けました。
MD simulations and Markov state models systematically characterized conformational landscapes of XBB.1, XBB.1.5 Omicron variants and their complexes showing that convergent mutation sites could control evolution allosteric pockets through modulation of conformational plasticity in the flexible adaptable regions.
MDシミュレーションとマルコフ状態モデルは、XBB.1、XBB.1.5オミクロン変異体とその複合体の立体構造ランドスケープを体系的に特徴付け、収束変異部位が柔軟な適応領域における立体構造可塑性の調節を通じて進化アロステリックポケットを制御できることを示した。
AlphaFold2-based structural modeling approaches were combined with all-atom MD simulations and mutational profiling of binding energetics and stability for prediction of dynamics, and binding of the SARS-CoV-2 Omicron BA.2.86 spike variant with ACE2 host receptor.
AlphaFold2ベースの構造モデリング手法は、全原子MDシミュレーションおよび結合エネルギーと安定性の変異プロファイリングと組み合わされ、SARS-CoV-2 Omicron BA.2.86スパイク変異体とACE2宿主受容体のダイナミクスと結合を予測しました。
This study quantified the role of the BA.2 and BA.2.86 backgrounds in modulating binding free energy changes revealing critical variant-specific contributions of the BA.2.86 mutational sites R403K, F486P and R493Q.
この研究では、結合自由エネルギーの変化を調節するBA.2およびBA.2.86バックグラウンドの役割を定量化し、BA.2.86変異部位R403K、F486P、およびR493Qの重要な変異体固有の寄与を明らかにしました。
AlphaFold predictions of multiple conformations with MD simulations identified important differences in the conformational landscapes and binding energetics of the XBB variants and revealing the mediating role of Q493 hotspot in epistatic couplings between L455F and F456L convergent mutations.
MDシミュレーションによる複数のコンフォメーションのAlphaFold予測により、XBB変異体のコンフォメーションランドスケープと結合エネルギーの重要な違いが特定され、L455FとF456L収束変異間のエピスタシス結合におけるQ493ホットスポットの媒介役割が明らかになりました。
The results of mutational scanning and binding analysis of the Omicron XBB spike variants with the ACE2 and a panel of class 1 antibodies provided a quantitative rationale to the experimental evidence that epistatic interactions of physically proximal binding hotspots Y501, R498, Q493, L455F and F456L residues can determine strong ACE2 binding, while convergent mutational sites F456L and F486P are instrumental in mediating broad antibody resistanc.
Omicron XBBスパイク変異体とACE2およびクラス1抗体パネルの変異スキャンおよび結合解析の結果は、物理的に近接した結合ホットスポットY501、R498、Q493、L455F、およびF456L残基のエピスタシス相互作用が強力なACE2結合を決定し、収束変異部位F456LおよびF486Pが広範な抗体耐性を媒介するのに役立つという実験的証拠に定量的な根拠を提供した。
We employed an integrative computational approach in which structure and conformational ensembles of the of the JN.1, KP.2 and KP.3 RBD-ACE2 complexes were first predicted using AlphaFold2 (AF2) methods using shallow multiple sequence alignment (MSA) approach.
我々は統合計算アプローチを採用し、まず浅い多重配列アラインメント (MSA) アプローチを用いた AlphaFold2 (AF2) 法を使用して、JN.1、KP.2、KP.3 RBD-ACE2 複合体の構造と立体配座の集合を予測しました。
The Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) approach is then employed for binding affinity computations of the Omicron RBD-ACE2 complexes.
次に、分子力学/一般化ボーン表面積 (MM-GBSA) アプローチを使用して、Omicron RBD-ACE2 複合体の結合親和性を計算しました。
We combined mutational profiling of the RBD residues with MM-GBSA approach for binding affinity computations of the RBD-ACE2 complexes across subvariants BA.2.86, JN.1, BA.2.86+F456L, BA.2.86+Q493E, BA.2.86+F456L/Q493E, BA.2.86+L455S/F456L (KP.1), BA.2.86+L455S/Q493E, KP.2 and BA.2.86+L455S/F456L/Q493E (KP.3).
RBD残基の変異プロファイリングとMM-GBSAアプローチを組み合わせて、サブバリアントBA.2.86、JN.1、BA.2.86+F456L、BA.2.86+Q493E、BA.2.86+F456L/Q493E、BA.2.86+L455S/F456L(KP.1)、BA.2.86+L455S/Q493E、KP.2、およびBA.2.86+L455S/F456L/Q493E(KP.3)にわたるRBD-ACE2複合体の結合親和性を計算しました。
Using mutational profiling of the S residues we identify binding energy hotspots and quantify epistatic couplings between convergent mutational hotspots.
S 残基の変異プロファイリングを使用して、結合エネルギー ホットスポットを特定し、収束変異ホットスポット間のエピスタシス結合を定量化します。
We examine a hypothesis that the emerging new variants may induce epistasis patterns where structural stability of the RBD can promote evolvability in vulnerable regions by tolerating combinations of convergent mutations at L455, F456, Q493 positions that confer beneficial phenotypes.
我々は、出現した新しい変異体が、有益な表現型をもたらす L455、F456、Q493 位置での収束変異の組み合わせを許容することで、RBD の構造安定性が脆弱な領域での進化を促進できるエピスタシス パターンを誘発する可能性があるという仮説を検証します。
Consistent with the latest functional studies of FLiRT and FLuQE variants, we found that that the Q493E mutation may be epistatically coupled with the F456L mutation, resulting in the reversal of the detrimental effect of Q493E seen in other backgrounds.
FLiRT および FLuQE 変異体の最新の機能研究と一致して、Q493E 変異は F456L 変異とエピスタシス的に結合し、他のバックグラウンドで見られる Q493E の有害な影響を逆転させる可能性があることがわかりました。
The results showed that combining the AF2 predictions conformational ensembles of RBD-ACE2 complexes with MM-GBSA computations of the RBD-ACE2 binding can produce robust quantitative analysis of binding mechanisms.
結果は、RBD-ACE2 複合体の AF2 予測コンフォメーション アンサンブルと RBD-ACE2 結合の MM-GBSA 計算を組み合わせることで、結合メカニズムの堅牢な定量分析を生成できることを示しました。
In addition, we also conducted mutational scanning of the S complexes with several important class I monoclonal antibodies to quantify the effect of convergent Omicron mutations on immune escape and the mechanism underlying balance between ACE2 binding and immune evasion of BA.2.86, JN.1, KP.2 and KP.3 variants.
さらに、いくつかの重要なクラス I モノクローナル抗体を使用して S 複合体の変異スキャンを実施し、収束性 Omicron 変異が免疫回避に与える影響と、BA.2.86、JN.1、KP.2、および KP.3 バリアントの ACE2 結合と免疫回避のバランスの基盤となるメカニズムを定量化しました。
The panel of studied antibodies included RBD-targeting antibodies such as S2K146, Omi-3 Omi-18, Omi-42 as well as antibodies BD55-5514 (SA55) and BD55-5840 (SA58) that bind to a different RBD epitope and are experimentally known to tolerate escape mutations in BA.2.8, JN.1, KP.2 and KP.3 variants.
研究対象となった抗体のパネルには、S2K146、Omi-3、Omi-18、Omi-42 などの RBD 標的抗体のほか、異なる RBD エピトープに結合し、実験的に BA.2.8、JN.1、KP.2、KP.3 バリアントのエスケープ変異を許容することが知られている抗体 BD55-5514 (SA55) および BD55-5840 (SA58) が含まれていました。
Structural analyses of BD-508, BD-236, BD-629, BD-604, and BD-515 that are class 1 antibodies and displayed a similar RBD-binding pose showed diverse interactions between antibodies and RBD and pointed to high diversity that RBD antibodies may exhibit.
クラス 1 抗体であり、類似の RBD 結合ポーズを示す BD-508、BD-236、BD-629、BD-604、および BD-515 の構造解析により、抗体と RBD の間に多様な相互作用が示され、RBD 抗体が示す可能性のある高い多様性が示されました。
We show that a group of convergent mutational sites Y453, L455 and F456 represent prevalent escape hotspots against class I antibodies as all modifications in these positions, particularly L455S and F456L, lead to a dramatic loss of binding interactions with the antibodies.
収束変異部位のグループである Y453、L455、および F456 は、クラス I 抗体に対する一般的なエスケープ ホットスポットを表していることを示しています。これらの位置、特に L455S と F456L のすべての変更により、抗体との結合相互作用が劇的に失われるからです。
A particular class of antibodies, F2 and F3 antibodies compete with ACE2, and their binding is affected by T376, K378, D405, R408 and G504, corresponding to class 1/4 which includes BD55-5514 (also known as SA55).
特定のクラスの抗体であるF2抗体とF3抗体はACE2と競合し、その結合は、BD55-5514(SA55としても知られる)を含むクラス1/4に対応するT376、K378、D405、R408、およびG504の影響を受けます。
Here, we demonstrate that SA55 and SA58 antibodies avoid targeting convergent mutational sites and therefore can overcome immune evasion.
ここでは、SA55 および SA58 抗体が収束変異部位を標的とすることを避け、免疫回避を克服できることを実証します。
The results suggest a mechanism in which convergent Omicron mutations can promote high transmissibility and antigenicity of the virus by controlling the interplay between the RBD stability and conformational adaptability, allowing for optimal fitness tradeoffs between binding to the host receptor and robust immune evasion profile.
結果は、収束オミクロン変異が RBD 安定性と立体構造適応性の相互作用を制御し、宿主受容体への結合と堅牢な免疫回避プロファイルの間の最適な適応トレードオフを可能にすることで、ウイルスの高い伝染性と抗原性を促進するメカニズムを示唆しています。
2. Materials and Methods
※省略します。
3. Results
3.1. Evolutionary and Phylogenetic Analysis of Differences between XBB, BA.2.86 and FLiRT Lineages
XBB、BA.2.86、FLiRT系統間の相違点の進化的および系統学的分析
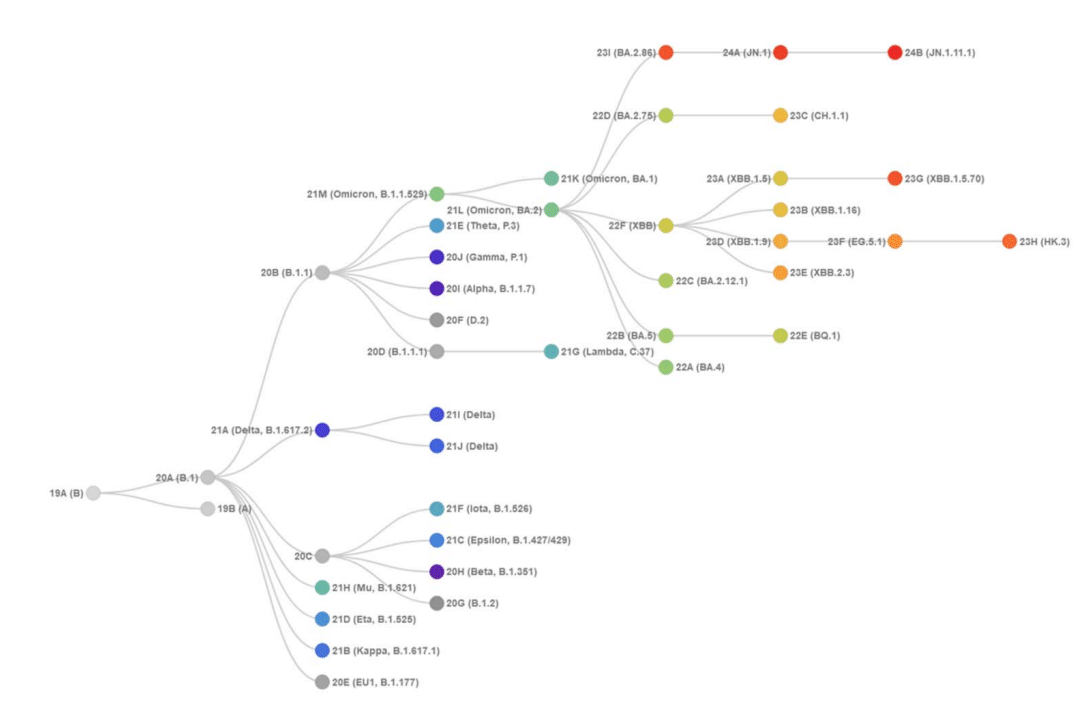
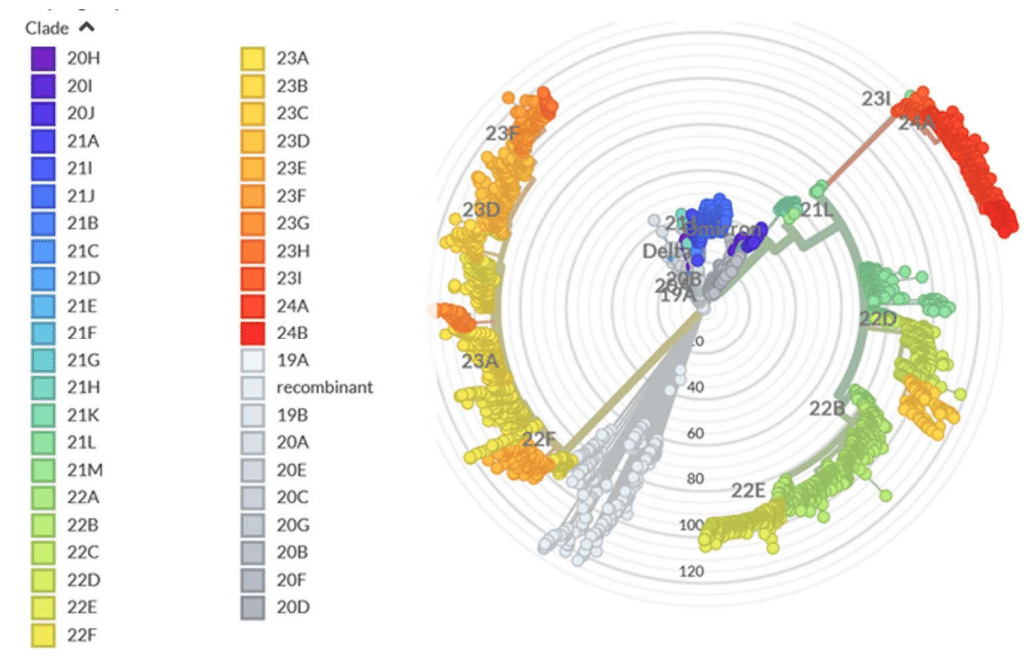
The evolutionary differences and divergence of XBB and BA.2.86 linages among the Omicron variants are illustrated by the phylogenetic analysis using their corresponding clades nomenclature from Nextstrain an open-source project for real time tracking of evolving pathogen populations (https://nextstrain.org/).
オミクロン変異体間の XBB および BA.2.86 系統の進化的差異と分岐は、進化する病原体集団をリアルタイムで追跡するオープンソース プロジェクトである Nextstrain の対応する系統名を使用した系統解析によって示されます。
Nextstrain provides dynamic and interactive visualizations of the phylogenetic tree of SARS-CoV-2, allowing users to explore the evolutionary relationships between different lineages and variants.
Nextstrain は、SARS-CoV-2 の系統樹を動的かつインタラクティブに視覚化して、ユーザーがさまざまな系統と変異体間の進化的関係を探索できるようにします。
This approach assigns SARS-CoV-2 variant as clade when it reaches a frequency of 20% globally at any time point.
このアプローチでは、SARS-CoV-2 変異体が、いつでも世界中で 20% の頻度に達した場合に、その変異体を系統群として割り当てます。
A new clade should be at least 2 mutations away from its parent major clade.
新しい系統群は、その親となる主要な系統群から少なくとも 2 つの変異が離れている必要があります。
According to the Nextstrain evolutionary analysis (Figures 2,3 )XBB.1.5 (23A clade) is a recombinant variant as it descends from XBB (22F clade).
Nextstrain の進化解析 (図 2、3) によると、XBB.1.5 (23A 系統) は XBB (22F 系統) の子孫であるため、組み換え変異体です。
XBB.1.5 has additional S mutations S:G252V and S:S486P that are also shared in XBB.1.5+F456L (EG.5) and XBB.1.5+L455F/F456L (XBB.1.5.70).
XBB.1.5 には、XBB.1.5+F456L (EG.5) および XBB.1.5+L455F/F456L (XBB.1.5.70) でも共有されている追加の S 変異 S:G252V および S:S486P があります。
The evolutionary analysis of different Omicron lineages indicated that BA.2.86 represents the step-change evolution that has given the virus a global growth advantage beyond the XBB.1-based lineages (Figures 2,3).
さまざまなオミクロン系統の進化分析により、BA.2.86 は、XBB.1 ベースの系統を超えてウイルスに世界的な成長の優位性を与えた段階的な進化を表していることが示されました (図 2、3)。
BA.2.86 S carries a novel and distinct constellation of mutations that produced a novel branch in the SARS-CoV-2 spike phylogenetic tree (Figures 2,3).
BA.2.86 S は、SARS-CoV-2 スパイク系統樹に新しい枝をもたらした、新しく独特な一連の変異を持っています (図 2、3)。
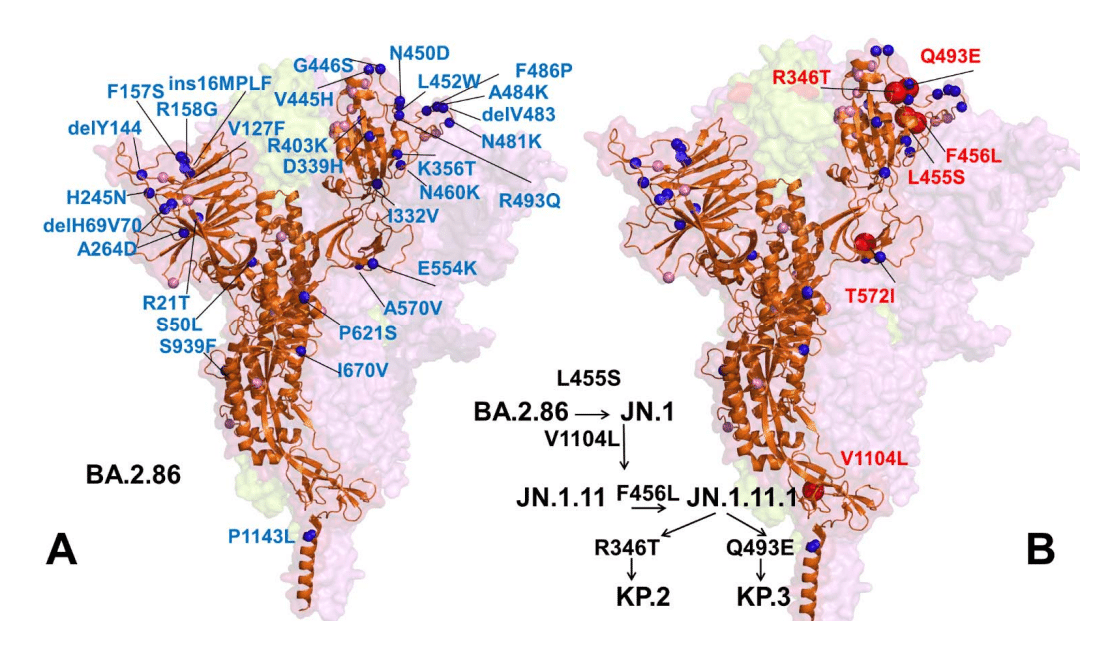
Compared with its BA.2 ancestor, BA.2.86 contains 34 mutations relative to BA.2 (29 substitutions, 4 deletions and 1 insertion) including RBD mutations I332V, D339H, K356T, R403K, V445H, G446S, N450D, L452W, N460K, N481K, delV483, A484K, F486P and R493Q (Figure 1).
BA.2 祖先と比較すると、BA.2.86 には BA.2 と比較して 34 の変異 (置換 29、欠失 4、挿入 1) が含まれており、RBD 変異 I332V、D339H、K356T、R403K、V445H、G446S、N450D、L452W、N460K、N481K、delV483、A484K、F486P、R493Q が含まれます (図 1)。
Many of these mutations such as G446S, N460K, F486P, and R493Q have been previously observed in other variants.
G446S、N460K、F486P、R493Q などのこれらの変異の多くは、以前に他の変異体で観察されています。
BA.2.86 consists of eight different sublineages, namely BA.2.86, BA.2.86.1, BA.2.86.2, BA.2.86.3, JQ.1, JN.3, JN.2 and JN.1.
BA.2.86 は、BA.2.86、BA.2.86.1、BA.2.86.2、BA.2.86.3、JQ.1、JN.3、JN.2、JN.1 の 8 つの異なる亜系統から構成されています。
JN.1 is a major descendent lineage of BA.2.86. JN.1 received Nextstrain clade classification 24A (Figures 2,3).
JN.1 は BA.2.86 の主要な子孫系統です。JN.1 は Nextstrain 系統分類 24A を受けました (図 2、3)。
In 2024 JN.1 diversified into a number of sublineages, many of which share recurrent mutations R346T ( JN.1.18), F456L ( JN.1.16), T572I ( JN.1.7), or combinations of these mutations (KP.2 variant).
2024 年に JN.1 はいくつかの亜系統に多様化し、その多くは反復変異 R346T (JN.1.18)、F456L (JN.1.16)、T572I (JN.1.7)、またはこれらの変異の組み合わせ (KP.2 変異体) を共有しています。
These mutations can individually contribute to the enhanced antibody evasion (R346T and F456L), ACE2-binding (R346T), and S protein stability and dynamic profile (T572I) when found on the genetic background of earlier Omicron subvariants.
これらの変異は、以前の Omicron 亜変異体の遺伝的背景で見つかった場合、強化された抗体回避 (R346T および F456L)、ACE2 結合 (R346T)、および S タンパク質の安定性と動的プロファイル (T572I) に個別に寄与する可能性があります。
The evolutionary diagrams show distinct evolutionary trajectories of SARS-CoV-2 Omicron XBB and BA.2.86/JN.1 lineages (Figure 3).
進化図は、SARS-CoV-2 オミクロン XBB 系統と BA.2.86/JN.1 系統の明確な進化の軌跡を示しています (図 3)。
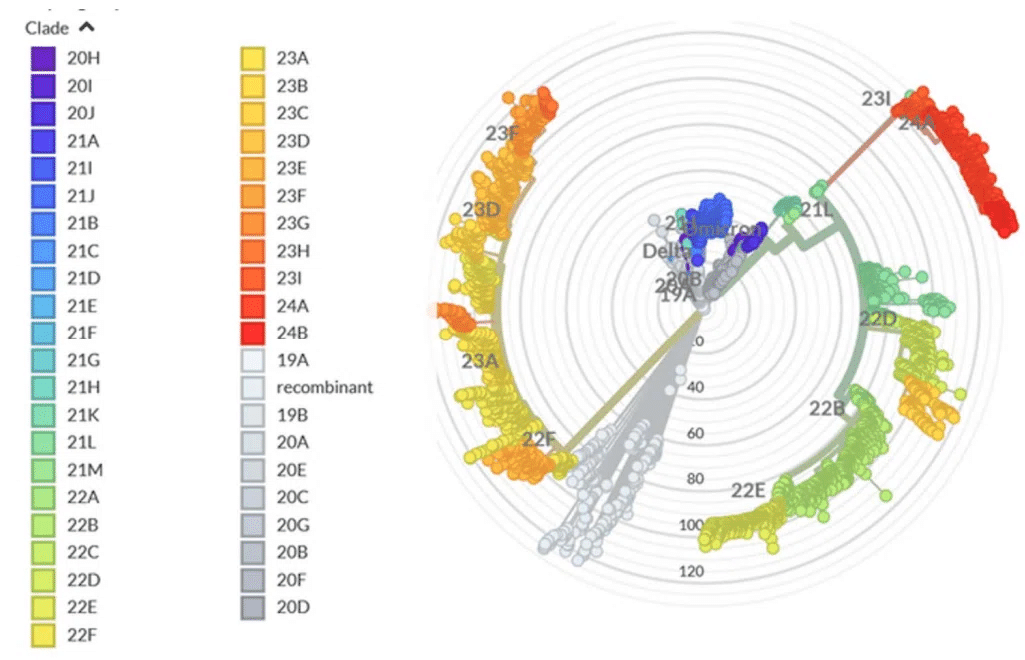
The current evolutionary divergences between XBB and BA.2.6, JN.1 lineages illustrated by Nextstrain diagrams (Figure 3) indicate that evolutionary trajectories of Omicron lineages can proceed through diverse mechanisms including complex recombination, antigenic drift and convergent evolution that led to convergent immune-escape mutations as many lineages independently acquired mutations such as R346T, L455F/S, F456L and T572I.
Nextstrain 図 (図 3) で示されている XBB 系統と BA.2.6、JN.1 系統間の現在の進化の分岐は、複雑な組み換え、抗原ドリフト、収束進化など、さまざまなメカニズムを通じてオミクロン系統の進化の軌跡が進む可能性があることを示しており、多くの系統が R346T、L455F/S、F456L、T572I などの変異を独立して獲得したため、収束免疫回避変異につながりました。
3.2 AF2-Based Modeling and Prediction of the BA.2.86, JN.1, KP.2 and KP.3 RBD-ACE2 Complexes and Conformational Ensembles
BA.2.86、JN.1、KP.2、KP.3 RBD-ACE2複合体およびコンフォメーションアンサンブルのAF2ベースのモデリングと予測
The lack of molecular details on structure, dynamics and binding energetics of the FLiRT and FLuQE variants including primarily JN.1, KP.2 and KP.3 RBD binding with the ACE2 receptor and antibodies provides a considerable challenge that needs to be addressed to rationalize the experimental data and establish the atomistic basis for the proposed molecular mechanisms.
FLiRT および FLuQE 変異体 (主に JN.1、KP.2、KP.3 RBD と ACE2 受容体および抗体との結合) の構造、ダイナミクス、結合エネルギーに関する分子の詳細が不足しているため、実験データを合理化し、提案された分子メカニズムの原子論的基礎を確立するには、対処する必要がある大きな課題があります。
These important objectives are addressed in the current study by using a combination of AI-based structural modeling approaches, MD simulations and the ensemble-based mutational profiling of the Omicron JN.1, KP.2 and KP.3 variants with the ACE2 receptor and a panel of monoclonal antibodies.
これらの重要な目的は、AI ベースの構造モデリング手法、MD シミュレーション、および ACE2 受容体とモノクローナル抗体のパネルを使用した Omicron JN.1、KP.2、KP.3 変異体のアンサンブルベースの変異プロファイリングを組み合わせて、現在の研究で対処されています。
We fist proceeded with AF2 structural predictions of the structures and conformational ensembles of the RBD-ACE2 complexes for the BA.2.86, JN.1, KP.2 and KP.3 variants using AF2 methodology with varied MSA depth.
まず、AF2 法を用いて、BA.2.86、JN.1、KP.2、KP.3 バリアントの RBD-ACE2 複合体の構造とコンフォメーション アンサンブルの AF2 構造予測を、MSA 深度を変えて実行しました。
By using two AF2 parameters in the following format: max_seqs:extra_seqs we can manipulate the number of sequences subsampled from the MSA (max_seqs sets the number of sequences passed to the row/column attention track and extra_seqs the number of sequences additionally processed by the main evoformer stack).
次の形式の 2 つの AF2 パラメータを使用することで、MSA からサブサンプリングされるシーケンスの数を操作できます (max_seqs は行/列アテンション トラックに渡されるシーケンスの数を設定し、extra_seqs はメインの evoformer スタックによって追加で処理されるシーケンスの数を設定します)。
More diverse predictions are generally encouraged with the lower values and MSA depth adaptations may facilitate conformational sampling.
一般的に、値が低いほど、より多様な予測が推奨され、MSA 深度適応によってコンフォメーション サンプリングが容易になる可能性があります。
The predicted structural ensembles for the BA.2.86, JN.1, KP.2 and KP.3 variants revealed a considerable heterogeneity of the RBD in all variants, showing a progressive increase in the mobility of the RBM region in KP.2 and KP.3 (Supporting Information, Figure S1).
BA.2.86、JN.1、KP.2、KP.3 バリアントの予測構造アンサンブルでは、すべてのバリアントで RBD のかなりの異質性が明らかになり、KP.2 と KP.3 では RBM 領域の可動性が徐々に増加していることが示されました (補足情報、図 S1)。
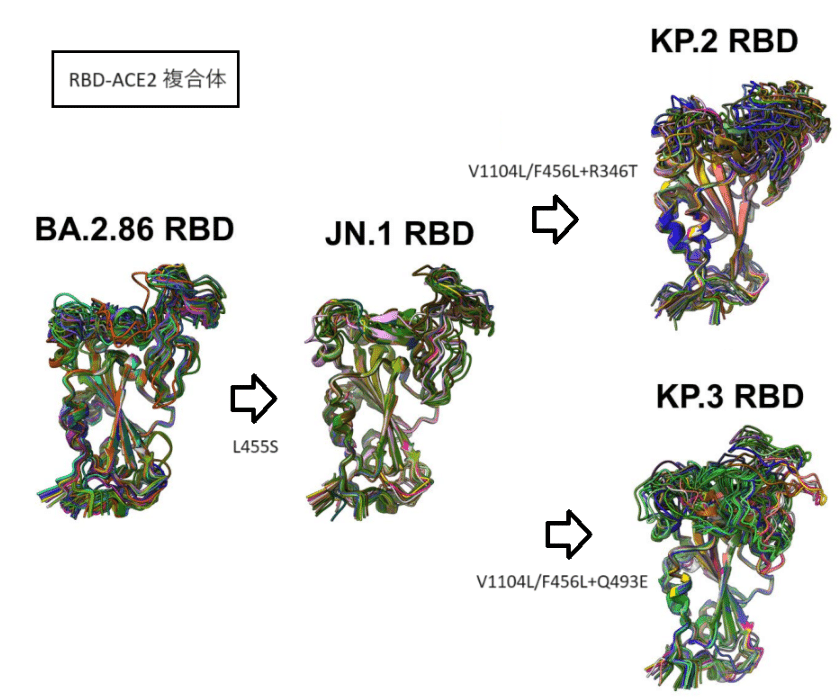
Interestingly, for BA.2.86 and JN.1 variants, structural variations in the RBD loop 444-452 are rather moderate but become more diffuse in the KP.2 and KP.3 variants (Supporting Information, Figure S1).
興味深いことに、BA.2.86 および JN.1 バリアントの場合、RBD ループ 444-452 の構造的変異は比較的中程度ですが、KP.2 および KP.3 バリアントではより拡散しています (補足情報、図 S1)。
The RBD loop regions 444-452 and 475-487 harbor BA.2.86 mutations V445H, G446S, N450D, L452W, delV483, A484K and F486P.
RBD ループ領域 444-452 および 475-487 には、BA.2.86 変異 V445H、G446S、N450D、L452W、delV483、A484K、F486P が含まれています。
These regions are also in the immediate structural proximity of L455, F456 and Q493 positions that undergo mutational changes in JN.1, KP.2 and KP.3 variants.
これらの領域は、JN.1、KP.2、および KP.3 バリアントで変異変化を受ける L455、F456、および Q493 位置の構造に近接しています。
The generated RBD conformation were evaluated using the pLDDT metric.
生成された RBD 構造は、pLDDT メトリックを使用して評価されました。
The distributions of pLDDT values displayed pronounced peaks at pLDDT ~70-85 for all studied variants (Figure 4).
pLDDT 値の分布は、調査したすべてのバリアントで pLDDT ~70-85 に顕著なピークを示しました (図 4)。
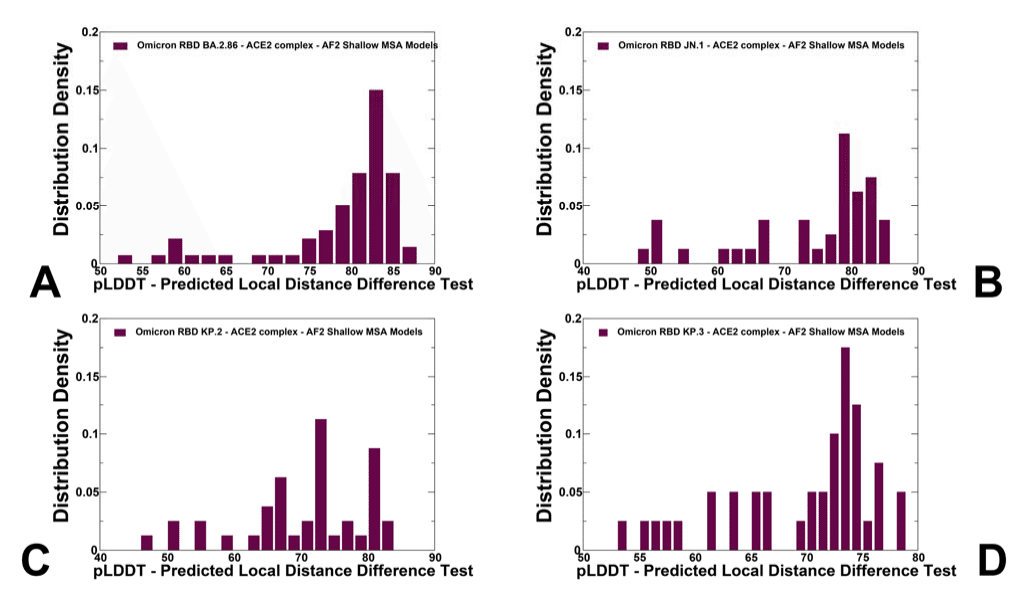
The pLDDT assessments of the structural ensembles for BA.2.86 variant (Figure 4A) showed that the dominant fraction of the ensemble (pLDDT ~80-85) conforms very closely with the experimental structure, while a minor fraction of the ensemble samples pLDDT values ~60- 80 associated with the native RBD conformations but featuring the increased mobility of the RBD flexible loops (Figure 4A).
BA.2.86 変異体に対する構造アンサンブルの pLDDT 評価 (図 4A) では、アンサンブルの主要部分 (pLDDT 約 80-85) が実験構造と非常によく一致しているのに対し、アンサンブル サンプルの少数部分は、ネイティブ RBD 構造に関連しますが、RBD フレキシブル ループの可動性が向上した pLDDT 値約 60-80 を示しています (図 4A)。
Although other pLDDT distributions were generally similar, we noticed more shallow profiles for JN.1 and KP.2 variants (Figure 4B,C).
他の pLDDT 分布は概ね同様でしたが、JN.1 および KP.2 変異体ではより浅いプロファイルが見られました (図 4B、C)。
A fairly significant number of smaller peaks at various pLDDT values (was seen for the KP.3 variant with a dominant distribution peak at pLDDT ~70-75 (Figure 4D).
KP.3 変異体では、さまざまな pLDDT 値でかなりの数の小さなピークが見られ、pLDDT 約 70-75 で主要な分布ピークが見られました (図 4D)。
AF2 predictions with pLDDT values ~70-90 are typically associated with high confidence while the regions with pLDDT values ~ 50-70 could indicate the reduced confidence.
pLDDT値が約70~90のAF2予測は通常、高い信頼性と関連しているが、pLDDT値が約50~70の領域は信頼性の低下を示している可能性がある。
Overall, the obtained conformational ensembles suggested the increased heterogeneity in the JN.1, KP.2 and especially KP.3 RBD variants which may potentially enable these variants to leverage a more mobile RBD structure to modulate and evade antibody neutralization.
全体的に、得られた立体構造集合は、JN.1、KP.2、特にKP.3 RBD変異体の不均一性の増加を示唆しており、これらの変異体がより可動性の高いRBD構造を利用して抗体中和を調節および回避できる可能性がある。
The distribution of structural similarity metric RMSD computed with respect to the cryo-EM structure of the BA.2.86 RBD-ACE2 complex echoed the pLDDT profile, showing strong peaks at low RMSD values ~ 1-2.5 Å for BA.2.86 (Figure 5A) and JN.1 variants (Figure 5B).
BA.2.86 RBD-ACE2複合体のクライオ電子顕微鏡構造に関して計算された構造類似性メトリックRMSDの分布はpLDDTプロファイルを反映し、BA.2.86(図5A)およびJN.1変異体(図5B)で約1〜2.5Åの低いRMSD値で強いピークを示した。
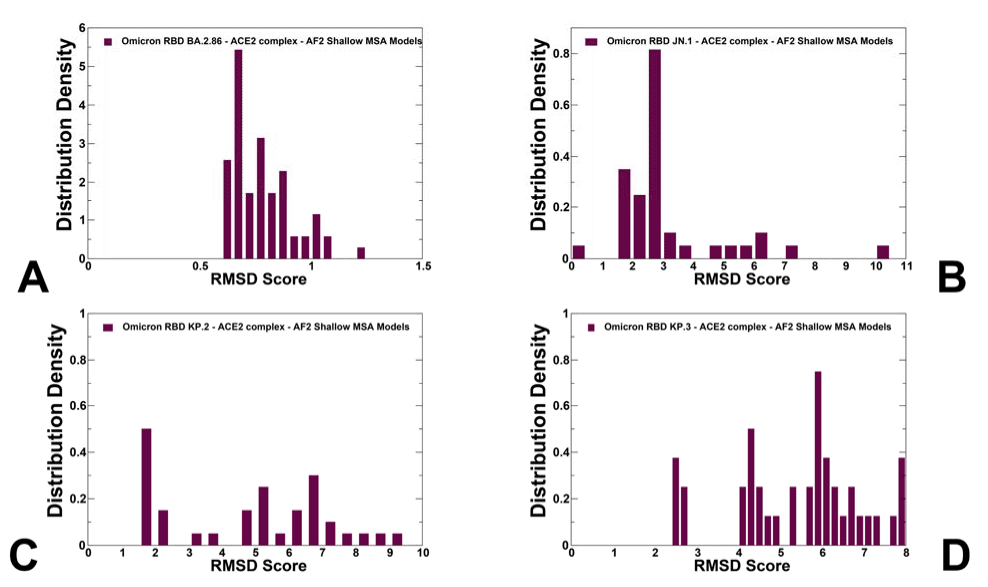
Although the profiles showed low RMSD peaks for KP.2 and KP.3 variants, these distributions also populated large RMSD values indicating the tendency of the AF2 predictions to produce overly heterogeneous conformations that could deviate significantly from the native structure of the BA.2.86 RBD (Figure 5C,D).
プロファイルは KP.2 および KP.3 バリアントの RMSD ピークが低いことを示しましたが、これらの分布には大きな RMSD 値も含まれており、AF2 予測が BA.2.86 RBD のネイティブ構造から大幅に逸脱する可能性のある過度に不均一なコンフォメーションを生成する傾向があることを示しています (図 5C、D)。
To select functionally relevant conformations from the ensemble, the predicted top models for the RBD-ACE2 complexes were selected solely based on the confidence metric pLDDT by choosing conformations with pLDDT > 70 (Figure 6).
アンサンブルから機能的に関連するコンフォメーションを選択するために、RBD-ACE2 複合体の予測されたトップモデルは、pLDDT > 70 のコンフォメーションを選択することで、信頼度メトリック pLDDT のみに基づいて選択されました (図 6)。
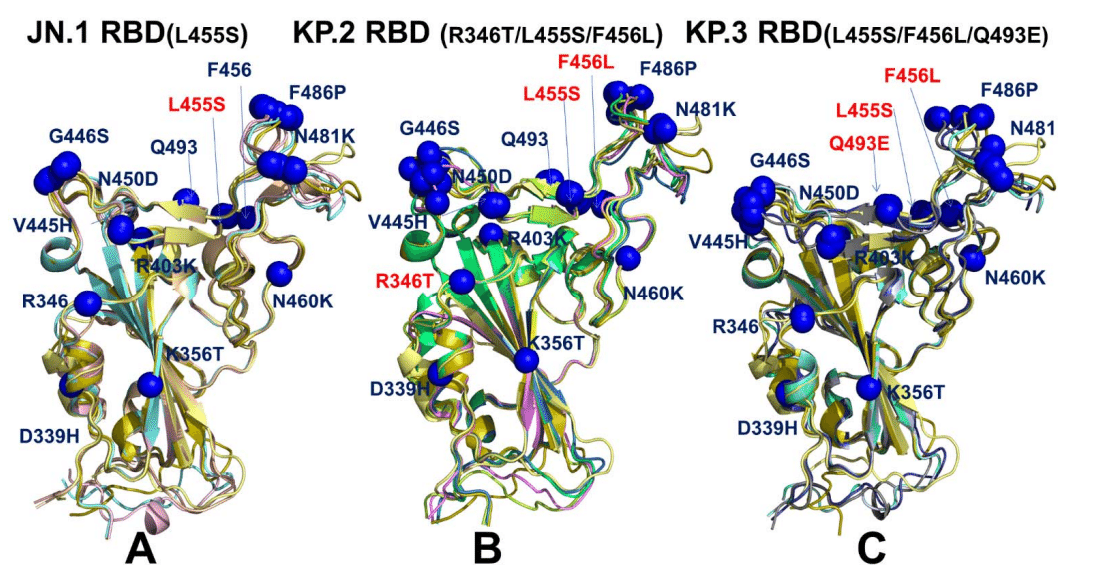
It is assumed that that conformations with high pLDDT values could be considered as functionally relevant representatives of the RBD mobility.
pLDDT 値が高い構造は、RBD 可動性の機能的に関連する代表として考えられると考えられます。
Structural alignment of the AF2 models with the recently released cryo-EM structure of the ancestor BA.2.86 RBD-ACE2 complex (pdb id 8QSQ) yielded the RMSD < 0.8 - 1.0 Å mostly displaying moderate deviations in the RBD loops which are especially apparent in the RBM region bearing mutations F486P and N481K (Figure 6).
AF2 モデルと、最近公開された祖先 BA.2.86 RBD-ACE2 複合体 (pdb id 8QSQ) のクライオ電子顕微鏡構造との構造アラインメントでは、RMSD < 0.8 - 1.0 Å が得られ、RBD ループに中程度の偏差がほとんど見られ、特に F486P および N481K 変異を伴う RBM 領域で顕著でした (図 6)。
The structural alignment of the predicted conformational ensembles (Figure 6) revealed only moderate heterogeneity of the flexible RBD loops while the RBD core remained largely rigid.
予測されたコンフォメーション アンサンブルの構造アラインメント (図 6) では、柔軟な RBD ループの異質性は中程度である一方、RBD コアはほぼ固定されたままであることが明らかになりました。
Moreover, structural analysis of the AF2-predicted ensemble conformations with high pLDDT value indicated that the degree of mobility in the RBD loops, particularly the RBM binding motif, can remain fairly similar across all variants.
さらに、pLDDT 値が高い AF2 予測アンサンブル コンフォメーションの構造解析では、RBD ループ、特に RBM 結合モチーフの可動性の程度は、すべてのバリアントでかなり類似していることがわかりました。
However, we noticed that JN.1 which is the immediate descendant of the BA.2.86 variant displayed very small deviations from the BA.2.86 cryo-EM structure (Figure 6A), including RBD loop 444-452.
しかし、BA.2.86 変異体の直系の子孫である JN.1 は、RBD ループ 444-452 を含め、BA.2.86 クライオ電子顕微鏡構造 (図 6A) から非常に小さな偏差を示していることに気付きました。
This is also evident from mapping of the mutational positions in all predicted conformations, showing that V445H, G446S, Q493, L455S, F456 sites remain virtually in the same positions in the top predicted conformations (Figure 6A).
これは、すべての予測されたコンフォメーションにおける変異位置のマッピングからも明らかであり、V445H、G446S、Q493、L455S、F456 サイトは、上位の予測されたコンフォメーションで実質的に同じ位置に留まっていることを示しています (図 6A)。
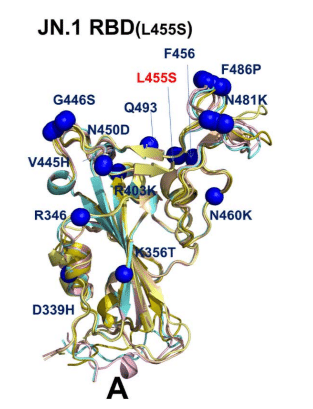
The alignment of JN.1 RBD conformations revealed only moderate plasticity of the RBM motif which are reflected in minor lateral displacements of F486P and N481K positions (Figure 6A).
JN.1 RBD コンフォメーションのアラインメントにより、RBM モチーフの可塑性は中程度であることが明らかになり、これは F486P および N481K 位置のわずかな横方向の変位に反映されています (図 6A)。
We observed a generally similar pattern in the KP.2 RBD conformations where moderate variability was seen in V445H/G446S sites, but the predicted positions of N481K and F486P remain largely the same despite the overall mobility of the RBM region (Figure 6B).
KP.2 RBD 構造でも、V445H/G446S 部位に中程度の変動が見られ、RBM 領域の全体的な可動性にもかかわらず、N481K と F486P の予測位置はほぼ同じままという、概ね同様のパターンが観察されました (図 6B)。
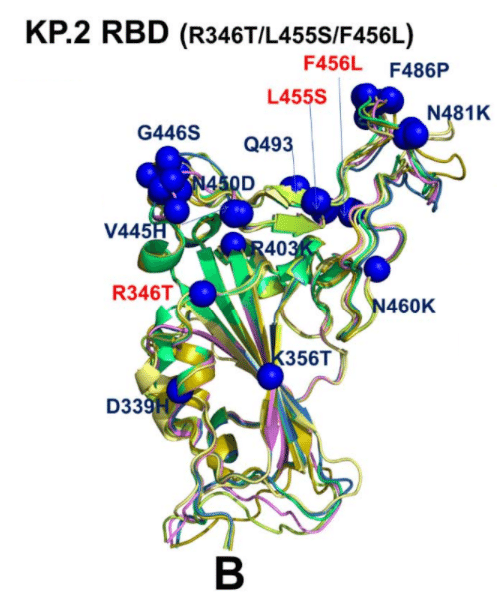
Interestingly, the RBD backbone conformations around L455S and F456L remains mainly rigid with only minor variations of the side-chains.
興味深いことに、L455SとF456L周辺のRBDバックボーン構造は、側鎖のわずかな変動のみで、主に剛性のままです。
Hence, functional heterogeneity of the RBD conformations may remain largely similar, suggesting that virus exploits convergent mutational sites to only moderately modulate ACE2 binding affinity, while the major role of these mutations is likely to enhance the immune evasion potential.
したがって、RBD 構造の機能的異質性はほぼ同様である可能性があり、ウイルスは収束変異部位を利用して ACE2 結合親和性を中程度に調整する一方、これらの変異の主な役割は免疫回避能力を高めることである可能性が高いことを示唆しています。
A greater conformational heterogeneity of the functional RBD conformations was observed in the KP.3 RBD-ACE2 complex (Figure 6C).
機能的 RBD 構造のより大きな構造異質性は、KP.3 RBD-ACE2 複合体で観察されました (図 6C)。
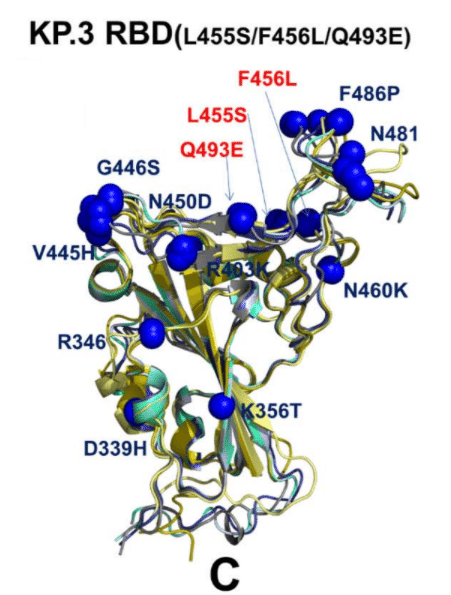
In this case, we found that the RBD loops 444-452 and 475-487 may display functionally relevant plasticity, where the RBB conformations and positions of F486P/N481K could slightly differ among top conformations (Figure 6C).
この場合、RBD ループ 444-452 および 475-487 が機能的に関連する可塑性を示す可能性があり、RBB 構造と F486P/N481K の位置はトップ構造間でわずかに異なる可能性があることがわかりました (図 6C)。
Although the majority of mutational sites remain in similar positions in different conformations of the ensemble, we found some moderate plasticity associated with convergent mutations R346T, L455S, F456L and Q493E as well as in their interacting ACE2 sites K31, H34, E35.
変異部位の大部分は、アンサンブルの異なる構造で同様の位置に留まりますが、収束変異 R346T、L455S、F456L、Q493E およびそれらの相互作用する ACE2 部位 K31、H34、E35 に関連する中程度の可塑性も見つかりました。
These findings from AF2 predictions suggest that L455S, F456L and Q493E in JN.1, KP.2 and KP.3 variants can undergo functional displacements around their native positions.
AF2 予測からのこれらの結果は、JN.1、KP.2、および KP.3 バリアントの L455S、F456L、および Q493E が、本来の位置の周囲で機能的に置換される可能性があることを示唆しています。
Accordingly, it would indicate that respective changes in the binding affinities with ACE2 may be partly affected by concerted conformational rearrangements at the binding interface.
したがって、ACE2 との結合親和性のそれぞれの変化は、結合界面での協調的な構造再配置によって部分的に影響を受ける可能性があることを示唆しています。
3.3 MD Simulations and Ensemble-Based Mutational Profiling of the Biding Interface Residues in the JN.1, KP.2 and KP.3 RBD-ACE2 Complexes
JN.1、KP.2、KP.3 RBD-ACE2複合体における結合界面残基のMDシミュレーションとアンサンブルベースの変異プロファイリング
We performed comparative all-atom MD simulations of the BA.2.86, JN.1, KP.2 and KP.3 RBD-ACE2 complexes.
BA.2.86、JN.1、KP.2、KP.3 RBD-ACE2 複合体の全原子 MD シミュレーションを比較しました。
The initial structure were taken from AF2 predicted ensembles.
初期構造は AF2 予測アンサンブルから取得しました。
We analyze MD simulations using a comparative analysis of the residue-based distance fluctuation stability indexes (Supporting Information, Figure S2).
残基ベースの距離変動安定性指標の比較分析を使用して MD シミュレーションを分析します (補足情報、図 S2)。
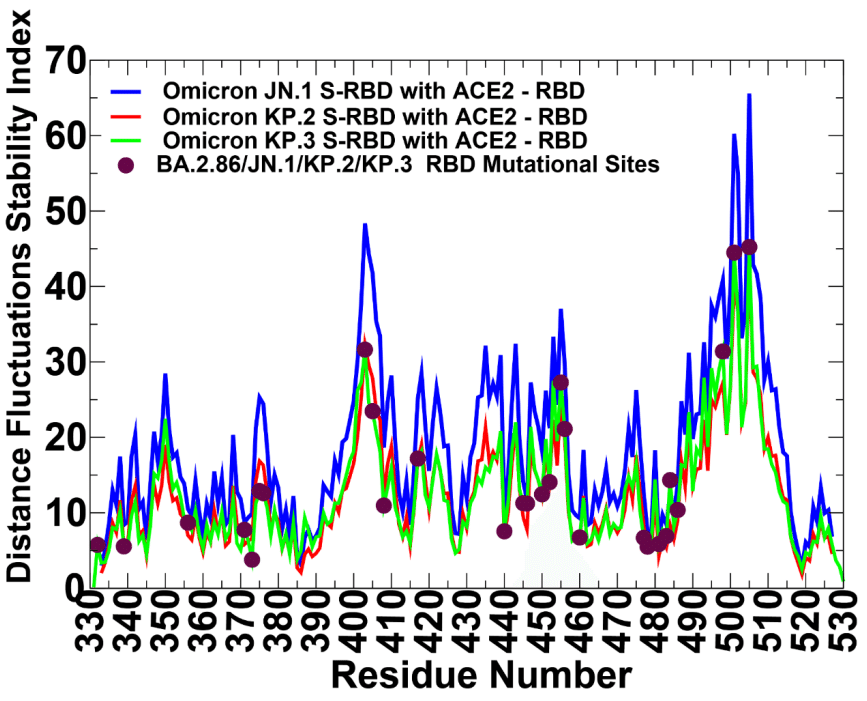
The distributions showed that the local maxima for all RBD-ACE2 complexes are aligned with structurally stable and predominantly hydrophobic regions in the RBD core (residues 400-406, 418-421, 453-456) as well as key binding interface clusters (residues 495-505) that include binding hotspots R498 and Y501.
分布から、すべての RBD-ACE2 複合体の極大値は、RBD コア内の構造的に安定で主に疎水性の領域 (残基 400-406、418-421、453-456) と、結合ホットスポット R498 および Y501 を含む主要な結合インターフェース クラスター (残基 495-505) に揃っていることが示されました。
The RBD positions associated with the high distance fluctuations stability indexes are F400, I402, Y421, Y453, L455, F456, Y473, A475, and Y489 (Supporting Information, Figure S2).
高い距離変動安定性指標に関連する RBD 位置は、F400、I402、Y421、Y453、L455、F456、Y473、A475、および Y489 です (補足情報、図 S2)。
The stability hotspots Y449, Y473, and Y489 are constrained by the requirements to maintain RBD stability and binding with the ACE2 host receptor.
安定性ホットスポット Y449、Y473、および Y489 は、RBD の安定性と ACE2 宿主受容体との結合を維持するための要件によって制約されています。
The profile minima are associated with the flexible RBD regions (residues 355-375, 381-394, 444-452, 455-471, 475-487).
プロファイルの最小値は、柔軟な RBD 領域 (残基 355-375、381-394、444-452、455-471、475-487) に関連しています。
Despite a similar shape of the distributions for all Omicron RBD variants, the larger peaks were seen for JN.1 distribution (Supporting Information, Figure S2).
すべての Omicron RBD バリアントの分布は同様の形状ですが、JN.1 分布ではより大きなピークが見られました (補足情報、図 S2)。
This implies that both the RBD core and ACE2-binding interface positions are more rigid in JN.1 and become somewhat more flexinle in KP.2 and especialy KP.3 variants.
これは、RBD コアと ACE2 結合インターフェースの位置はどちらも JN.1 ではより硬く、KP.2 および特に KP.3 バリアントではやや柔軟になっていることを意味します。
The profiles pointed to flexible RBD regions (residues 355-375, 381-394, 444-452, 455-471, 475-487) that are shared by all variants and are associated with low stability indexes.
プロファイルは、すべてのバリアントに共通し、低い安定性指数に関連付けられている柔軟な RBD 領域 (残基 355-375、381-394、444-452、455-471、475-487) を示しました。
Of particular interest are the lowered distance fluctuation stability indexes in the RBD regions 440-460 and 470-487 for KP.2 and KP.3 (Supporting Information, Figure S2).
特に興味深いのは、KP.2 および KP.3 の RBD 領域 440-460 および 470-487 で距離変動安定性指数が低下していることです (補足情報、図 S2)。
These observations are consistent with AF2 predictions suggesting a generlly more flexible RBD in the KP.2/KP.3 RBD variants.
これらの観察結果は、KP.2/KP.3 RBD バリアントでは RBD が一般的に柔軟であることを示唆する AF2 予測と一致しています。
By mapping BA.2.86 mutational sites on the distriutions, we noticed that mutated RBD positions are typicaly characterized by moderate stability indexes, indicating that Omicron mutations target conformationally adaptable regions in the RBD.
BA.2.86 変異部位を分布にマッピングすると、変異した RBD 位置は典型的には中程度の安定性指数で特徴付けられ、オミクロン変異が RBD 内の構造的に適応可能な領域をターゲットにしていることがわかりました。
Mutational positions N440K, V445P, G446S, N460K, F486P displayed low distance fluctuation stability indexes, which may indicate presence of local mobility in these regions.
変異位置 N440K、V445P、G446S、N460K、F486P は、低い距離変動安定性指数を示しており、これらの領域に局所的な可動性が存在することを示している可能性があります。
Several important RBD binding interface centers L455 and F456 showed intermediate stability indexes, indicanting that although these positions remain overall stable in all variants, these sites may undero moderate fluctuations in their mutational forms containing L455S and F456L changes (Supporting Information, Figure S2).
いくつかの重要な RBD 結合インターフェース センター L455 と F456 は中程度の安定性指数を示しており、これらの位置はすべてのバリアントで全体的に安定しているものの、L455S と F456L の変化を含む変異型では、これらのサイトが中程度の変動を受ける可能性があることを示しています (補足情報、図 S2)。
At the same time, R498, Y501 and H505 featured high stability indexes, reflecting a considerable rigidification of these residues due to strong interactions with ACE2.
同時に、R498、Y501、H505 は高い安定性指数を示し、ACE2 との強い相互作用によりこれらの残基がかなり硬直化したことを反映している。
We fist examined in some detail the predicted binding interfaces in the JN.1, KP.2 and KP.3 complexes.
まず、JN.1、KP.2、KP.3 複合体の予測される結合界面を詳細に調べました。
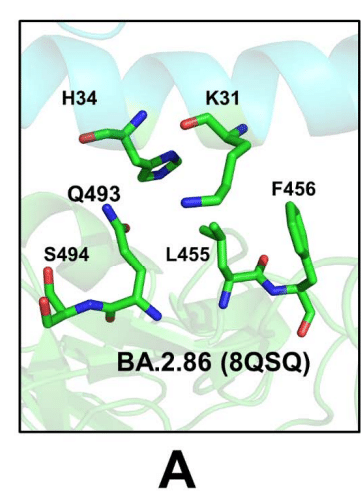
The binding interface in the cryo-EM structure of the BA.2.86 RBD-ACE2 complex (pdb id 8QSQ) showed that positions of H34 and K31 ACE2 residues as well as RBD residues Q493, L455 and F456 are very similar to those in the XBB.1.5 RBD-ACE2 complex (Figure 7A, Supporting Information, Figure S3).
BA.2.86 RBD-ACE2 複合体 (pdb id 8QSQ) のクライオ電子顕微鏡構造における結合界面は、H34 および K31 ACE2 残基、ならびに RBD 残基 Q493、L455、および F456 の位置が、XBB.1.5 RBD-ACE2 複合体のものと非常に類似していることを示しました (図 7A、補足情報、図 S3)。
In the XBB.1.5 RBD-ACE2 complex (pdb id 8WRL) K31 side chain is placed between Q493 and hydrophobic L455 and F456 residues, showing that movements of H34 and K31 are fairly restricted (Supporting Information, Figure S3A).
XBB.1.5 RBD-ACE2 複合体 (pdb id 8WRL) では、K31 側鎖が Q493 と疎水性 L455 および F456 残基の間に配置されており、H34 および K31 の動きがかなり制限されていることを示しています (補足情報、図 S3A)。
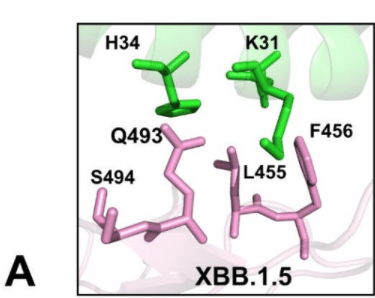
A similar interfacial pattern was predicted for XBB.1.5 +L455F mutant where the favorable position of Q493 remained largely unchanged as compared to XBB.1.5 (Supporting Information, Figure S3B).
XBB.1.5 +L455F変異体についても同様の界面パターンが予測され、Q493の好ましい位置はXBB.1.5と比較してほとんど変化しませんでした(補足情報、図S3B)。
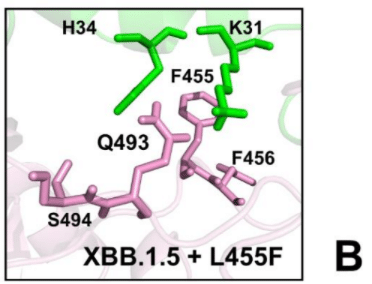
Compared to XBB.1.5, F456L mutation does not substantially affect the interactions on the RBD-ACE2 interface and yet Q493 and H34 undergo some rearrangements due to the increased conformational space which allows insertion of H34 side chain between Q493 and S494 (Supporting Information, Figure S3C).
XBB.1.5と比較して、F456L変異はRBD-ACE2界面の相互作用に実質的に影響しませんが、Q493とH34は、Q493とS494の間にH34側鎖を挿入できるコンフォメーションスペースの増加により、いくらかの再配置を受けます(補足情報、図S3C)。
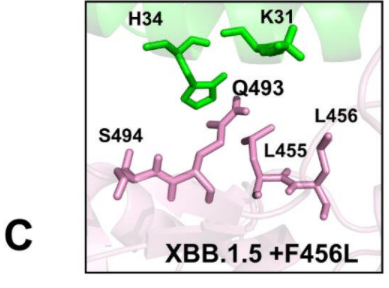
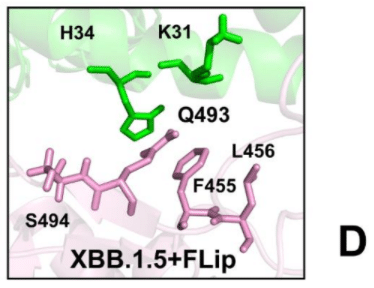
A similar interfacial pattern is further strengthened in the XBB.1.5 +FLip RBD-ACE2 complex (pdb id 8WRH) [111] the flip of L455 and F456 provided synergistic reorganization of the interactions between H34 and Q493/S494 RBD residues (Supporting Information, Figure S3D).
同様の界面パターンは XBB.1.5 +FLip RBD-ACE2 複合体 (pdb id 8WRH) でもさらに強化され、L455 と F456 の反転により、H34 と Q493/S494 RBD 残基間の相互作用が相乗的に再編成されました (補足情報、図 S3D)。
We examined the details of the predicted binding interface in the vicinity of L455 and F456 positions to illustrate the effect of mutations of L455 and F456 on the structural repacking induced by the L455F/F456L mutation and change in the binding contacts.
L455 と F456 の位置付近で予測される結合界面の詳細を調べ、L455 と F456 の変異が L455F/F456L 変異によって誘発される構造再パッキングと結合接触の変化に与える影響を明らかにしました。
Of special interest is the analysis of the binding interface in the KP.3 RBD-ACE2 complex, primarily to examine whether Q493E mutation can induce potential cooperative structural changes that may result in the observed epistatic improvement in the ACE2 binding affinity.
特に興味深いのは、KP.3 RBD-ACE2 複合体の結合界面の分析です。主に、Q493E 変異が、ACE2 結合親和性の観察されたエピスタシス改善につながる可能性のある潜在的な協調的構造変化を誘発できるかどうかを調べるためです。
Q493E involves the rarest of all nucleotide mutations, C->G, and occurs at a key residue.
Q493E は、すべてのヌクレオチド変異の中で最もまれな C->G を伴い、重要な残基で発生します。
Intriguingly, Q493E marks another major change by reversing the basic trend in which the RBD interfacial region (438-506) has more basic i.e. positively charged in most of the Omicron variants.
興味深いことに、Q493E は、ほとんどの Omicron バリアントで RBD 界面領域 (438-506) がより塩基性、つまり正に帯電しているという基本的な傾向を逆転させることで、もう 1 つの大きな変化を示しています。
In fact, with V445H, N481K, and A484K mutations in BA.2.86 this variant exemplified largest increase in the positively charged RBD sites.
実際、BA.2.86 の V445H、N481K、および A484K 変異により、このバリアントは正に帯電した RBD サイトの最大の増加を示しました。
Quite unexpectedly, Q493E, in the KP.3 variant reverses this trend, making the RBM more acidic.
まったく予想外なことに、KP.3 変異体の Q493E はこの傾向を逆転させ、RBM をより酸性にします。
We leveraged the conformational ensembles obtained from AF2 and MD simulations to map the most favorable interfacial interactions in the KP.3 RBD-ACE2 complex (Figure 7B).
AF2 および MD シミュレーションから得られたコンフォメーション アンサンブルを活用して、KP.3 RBD-ACE2 複合体における最も好ましい界面相互作用をマッピングしました (図 7B)。
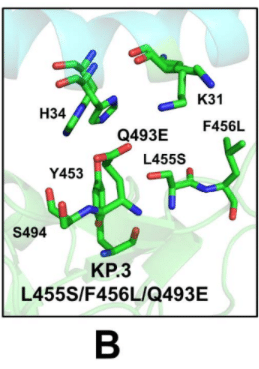
The second region of the RBD that includes important mutational positions R403, L455, Y453, and Q493 binds with the central segment of the α1 helix interface of the ACE2 interface residues (D30, K31, H34, E35 and D38).
重要な変異位置 R403、L455、Y453、および Q493 を含む RBD の 2 番目の領域は、ACE2 インターフェース残基 (D30、K31、H34、E35、および D38) の α1 ヘリックス インターフェースの中央セグメントと結合します。
Structural analysis showed that both H34 and K31 ACE2 residues become more flexible in the complex as compared to XBB.1.5 and BA.2.86 complexes (Figure 7B).
構造解析により、H34 および K31 ACE2 残基はどちらも、XBB.1.5 および BA.2.86 複合体と比較して複合体内でより柔軟になることが示されました (図 7B)。
Moreover, we found that H34 and K31 can assume several preferential side-chain orientations that enable moderate structural rearrangements which allow more room for making stable hydrogen bond interactions with Q493Q (Figure 7B).
さらに、H34 と K31 は、Q493Q との安定した水素結合相互作用を形成するための余地を増やす、中程度の構造再配置を可能にするいくつかの優先的な側鎖配向をとることができることがわかりました (図 7B)。
In addition, it is observed that the H34 side-chain placements allow for stronger contacts with Y453 residue.
さらに、H34 側鎖の配置により、Y453 残基とのより強い接触が可能になることが観察されています。
The additional comparative analysis illustrated superposition of the predicted binding interface for KP.3 RBD-ACE2 complex with the recently determined structures of the BA.4/5 RBD-ACE2 complex (pdb id 8H06) (Figure 7C).
追加の比較分析により、KP.3 RBD-ACE2複合体の予測される結合界面と、最近決定されたBA.4/5 RBD-ACE2複合体(pdb id 8H06)の構造との重ね合わせが示されました(図7C)。
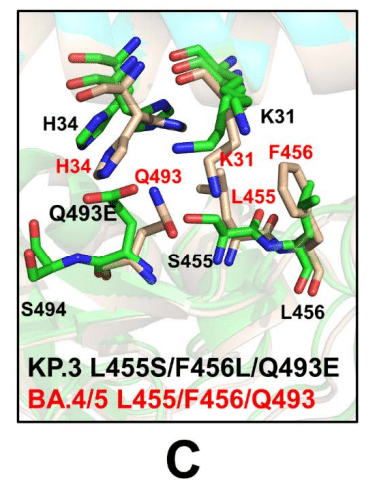
Interestingly, the side chain of H34 adopts two alternative conformations in the BA.4/5 complex that are essentially identical to the preferential side-chain conformation of H34 in the KP.3 complex.
興味深いことに、BA.4/5複合体のH34の側鎖は、KP.3複合体のH34の優先側鎖コンフォメーションと本質的に同一である2つの代替コンフォメーションを採用しています。
H34 of hACE2 forms hydrogen bonding with Y453 of the BA.4/5 RBD and Q493 is hydrogen-bonded to K31.
hACE2のH34は、BA.4/5 RBDのY453と水素結合を形成し、Q493はK31と水素結合しています。
Similarly, the predicted K31 side-chain positions in the KP.3 complex are very similar to the experimentally determined K31 orientation in the BA.4/5 complex (Figure 7C).
同様に、KP.3複合体の予測されたK31側鎖の位置は、実験的に決定されたBA.4/5複合体のK31の向きと非常に似ています(図7C)。
Hence, structural analysis of the ensemble-predicted binding interface for the KP.3 complex suggested the moderately increased plasticity of the ACE2 interfacial sites that are afforded by L455S/F456L mutations that reduced bulkiness of the hydrophobic packing and conferred room for the increased flexibility and improved interactions mediated by Q493E with H34 and K31 hotspots (Figure 7C).
したがって、アンサンブル予測による KP.3 複合体の結合界面の構造解析から、ACE2 界面部位の可塑性が中程度に増加していることが示唆されました。これは、L455S/F456L 変異によって疎水性パッキングのかさばりが軽減され、柔軟性が高まり、Q493E と H34 および K31 ホットスポットとの相互作用が改善される余地が生まれたことによるものです (図 7C)。
In addition, we found that Q493 lies in the structural proximity of RBD residues 447-457, 467-473, 484-484, 488-497.
さらに、Q493 は RBD 残基 447-457、467-473、484-484、488-497 の構造的近傍にあることがわかりました。
In particular, Q493E is close to residues 346 (R346T in FLiRT variants), 448-456, and the 483-484 region where BA.2.86 has ∆V483 and A484K.
特に、Q493E は残基 346 (FLiRT バリアントの R346T)、448-456、および BA.2.86 に ∆V483 と A484K がある 483-484 領域に近いです。
Using conformational ensembles obtained from MD simulation studies of the RBD-ACE2 complexes for the BA.2.86, JN.1, KP.2 and KP.3 variants we performed a systematic mutational scanning of the RBD residues in the RBD-ACE2 complexes.
BA.2.86、JN.1、KP.2、KP.3 変異体の RBD-ACE2 複合体の MD シミュレーション研究から得られたコンフォメーション アンサンブルを使用して、RBD-ACE2 複合体の RBD 残基の系統的な変異スキャンを実行しました。
Mutational scanning of the key RBD positions in the cryo-EM structure of the BA.2.86 RBD-ACE2 complex (Supporting Information, Figure S4) was analyzed focusing on the RBD positions that correspond to sites of convergent evolution and determine changes seen in JN.1 (L455S), KP.2 (R346T, L455S, F456L) and KP.3 (L455S, F456L, Q93E).
BA.2.86 RBD-ACE2 複合体のクライオ電子顕微鏡構造 (補足情報、図 S4) における主要な RBD 位置の変異スキャンは、収斂進化の部位に対応し、JN.1 (L455S)、KP.2 (R346T、L455S、F456L)、KP.3 (L455S、F456L、Q93E) に見られる変化を決定する RBD 位置に焦点を当てて分析されました。
Most of mutations produced large destabilization binding free energy values with the exception of Y453F that resulted in the negligible change (Supporting Information, Figure S4A).
変異のほとんどは、大きな不安定化結合自由エネルギー値を生み出しましたが、Y453F は例外で、変化は無視できるものでした (補足情報、図 S4A)。
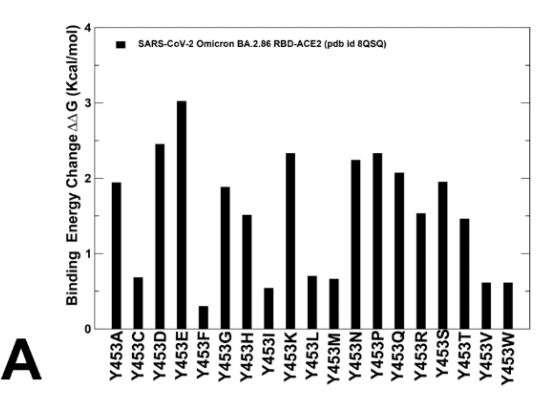
This is in full agreement with the experimental studies that demonstrated role of Y453F as an adaptive mutation which increased virus interaction with mink ACE2 receptor, without compromising its utilization of human ACE2 receptor.
これは、Y453F が適応変異としての役割を実証した実験研究と完全に一致しており、この変異は、ヒト ACE2 受容体の利用を損なうことなく、ミンク ACE2 受容体とのウイルスの相互作用を増加させました。
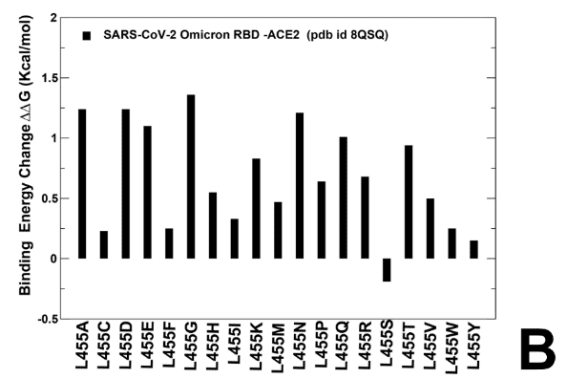
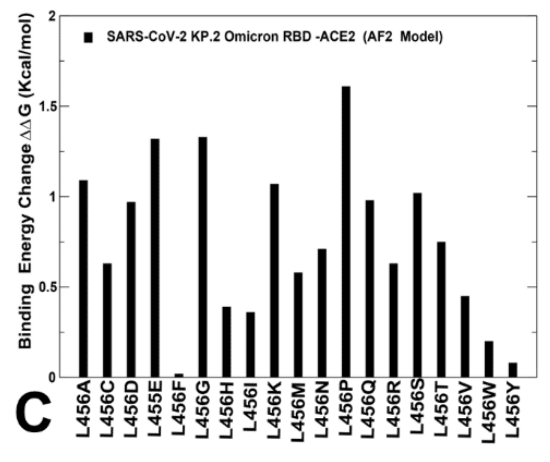
Mutational profiling at L455 showed that all modifications, particularly L455S are appreciably deleterious for ACE2 binding (ΔΔG ~ 1.0 kcal/mol) (Supporting Information, Figure S4B).
L455 での変異プロファイリングでは、すべての変更、特に L455S が ACE2 結合にかなり有害であることが示されました (ΔΔG 約 1.0 kcal/mol) (補足情報、図 S4B)。
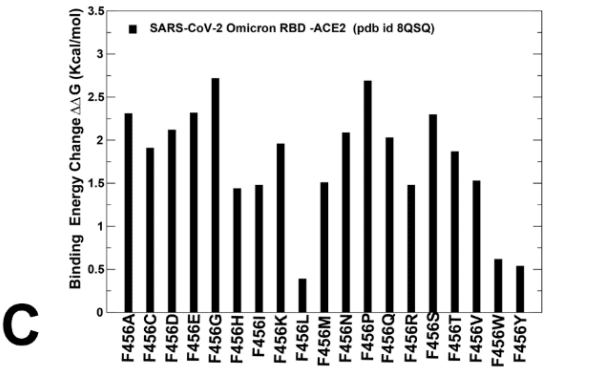
Mutational changes at F456 position of the BA.2.86 RBD-ACE2 structure are generally very unfavorable leading to ΔΔG ~ 1.5-2.0 kcal/mol.
BA.2.86 RBD-ACE2 構造の F456 位置での変異変化は、一般的に非常に不利であり、ΔΔG 約 1.5-2.0 kcal/mol につながります。
However, F456L mutational change alone in the genetic background of BA.2.86 is rather small ( ΔΔG ~ 0.3 kcal/mol ) and within the error margin, indicating that even individually F456L cannot significantly decrease the ACE2 binding affinity (Supporting Information, Figure S4C).
ただし、BA.2.86 の遺伝的背景における F456L 変異変化のみはかなり小さく (ΔΔG 約 0.3 kcal/mol)、誤差範囲内であるため、F456L 単独でも ACE2 結合親和性を大幅に低下させることはできないことが示されています (補足情報、図 S4C)。
Of particular interest are mutations in Q493 position showing relatively moderate destabilization for Q493R and Q493E ( ΔΔG ~ 0.6 kcal/mol) (Supporting Information, Figure S4D).
特に興味深いのは、Q493R と Q493E の比較的中程度の不安定化を示す Q493 位置の変異です (ΔΔG 約 0.6 kcal/mol) (補足情報、図 S4D)。
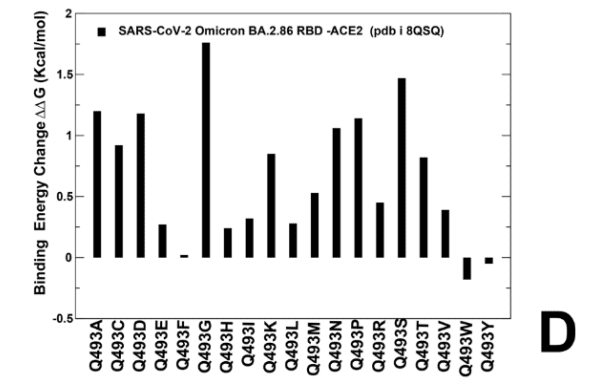
These results agree with DMS studies that projected Q493E as destabilizing mutation in the backgrounds of XBB.1.5 variant.
これらの結果は、XBB.1.5 変異体のバックグラウンドで Q493E を不安定化変異と予測した DMS 研究と一致しています。
Our results suggested that F456L and Q493E may be only marginally deleterious and may reverse their effect on ACE2 binding.
私たちの結果は、F456L と Q493E はわずかに有害であり、ACE2 結合に対する効果を逆転させる可能性があることを示唆しています。
We then leveraged AF2 and MD conformational ensembles to perform ensemble-based mutational scanning of key RBD positions that are mutated in the JN.1, KP.2 and KP.3 (Figure 8).
次に、AF2 および MD 立体構造アンサンブルを利用して、JN.1、KP.2、および KP.3 で変異している主要な RBD 位置のアンサンブルベースの変異スキャンを実行しました (図 8)。
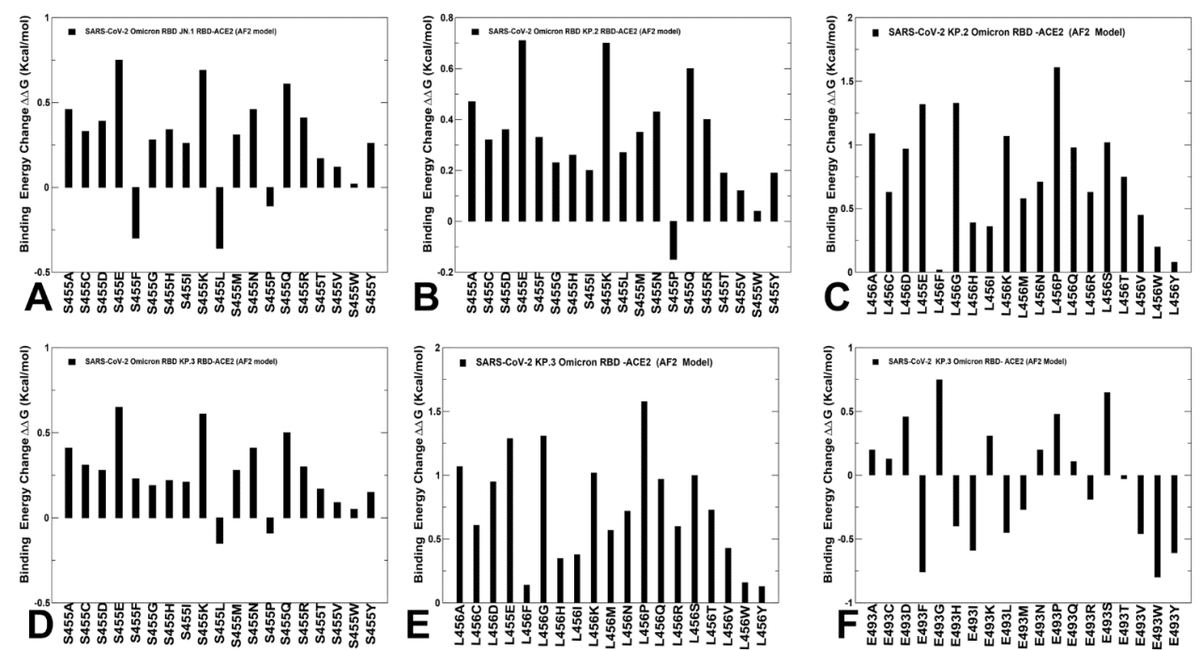
Mutational analysis of S455 in the JN.1 RBD-ACE2 ensemble showed that mutations S455F, S455L and S455P are favorable and the reversed S455L change can potentially lead to some improvement in binding affinity (Figure 8A).
JN.1 RBD-ACE2 アンサンブルにおける S455 の変異解析により、変異 S455F、S455L、および S455P は有利であり、S455L の逆転は結合親和性のいくらかの改善につながる可能性があることが示されました (図 8A)。
This further confirmed that L455S mutation can markedly decrease ACE2 binding which is consistent with the experimental data.
これにより、L455S 変異は ACE2 結合を著しく低下させる可能性があることがさらに確認され、これは実験データと一致しています。
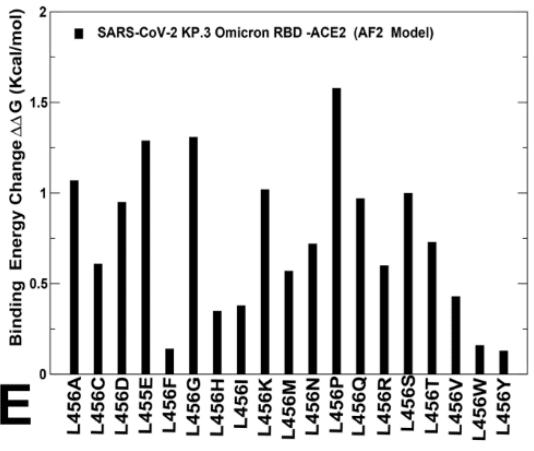
We then investigated more closely the effect of mutations in key sites of KP.2 (R346T/L455S/F456L) KP.3 variant (S455, L456 and E493).
次に、KP.2 (R346T/L455S/F456L) KP.3 変異体の主要部位 (S455、L456、E493) における変異の影響をさらに詳しく調査しました。
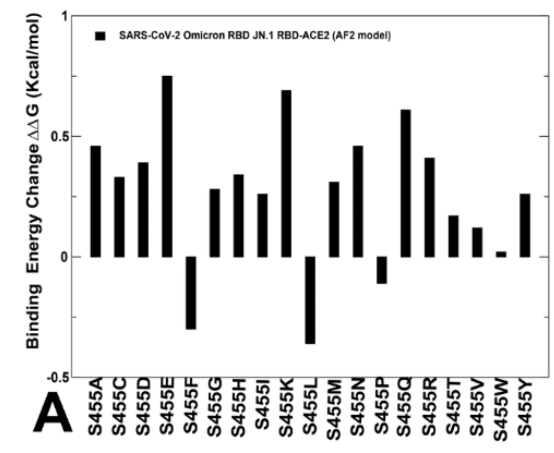
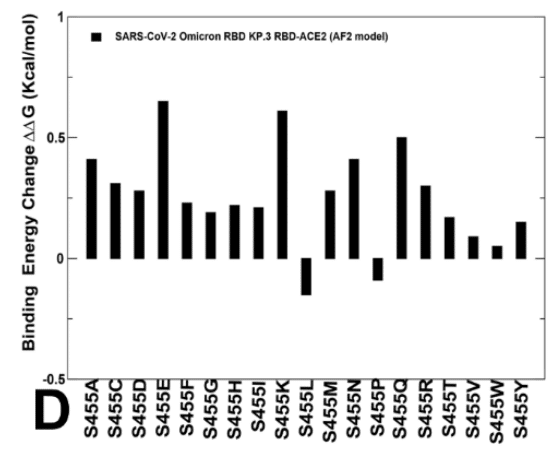
The results showed that S455F, S455L and S455P remain favorable which implies that L455S substitution is deleterious for ACE2 binding in different genetic backgrounds of JN.1 and KP.3 (Figure 8B,D).
結果は、S455F、S455L、S455P が依然として好ましいことを示し、これは L455S 置換が JN.1 と KP.3 の異なる遺伝的背景における ACE2 結合に有害であることを示唆しています (図 8B、D)。
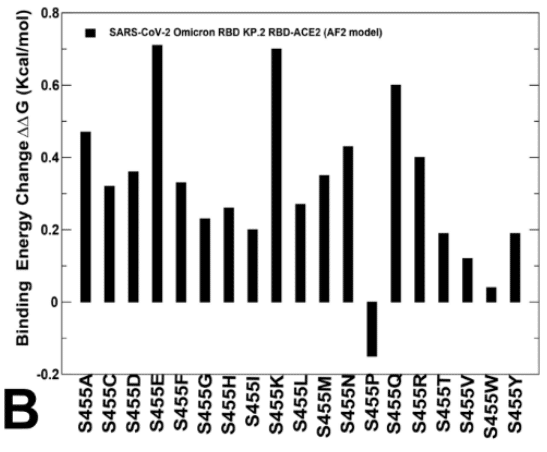
Moreover, the L456 position in both KP.2 and KP.3 RBD-ACE2 complexes appears to be highly favorable as all substitutions lead to destabilizing ΔΔG ~ 0.5-2.0 kcal/mol (Figure 8C,E).
さらに、KP.2 と KP.3 の両方の RBD-ACE2 複合体における L456 の位置は、すべての置換が ΔΔG を約 0.5-2.0 kcal/mol 不安定化させるため、非常に好ましいようです (図 8C、E)。
The positive binding free energy values ΔΔG correspond to destabilizing changes and negative binding free energy changes are associated with stabilizing changes.
正の結合自由エネルギー値 ΔΔG は不安定化変化に対応し、負の結合自由エネルギー変化は安定化変化と関連しています。
Mutational profiling of E493 in the KP3 RBD-ACE2 complex showed a more complex and diverse picture as a number of modifications can improve binding affinity (Figure 8F) which is consistent with the fact that E493 is generally destabilizing change.
KP3 RBD-ACE2 複合体における E493 の変異プロファイリングでは、いくつかの変更によって結合親和性が向上するため、より複雑で多様な画像が示されました (図 8F)。これは、E493 が一般に不安定化変化であるという事実と一致しています。
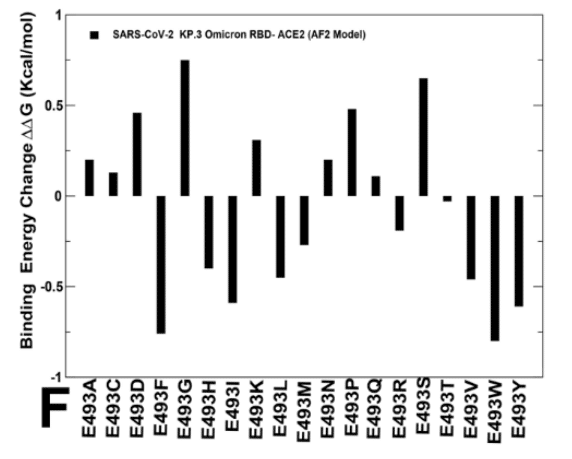
However, in the KP.3 background, the reverse mutation E493Q appeared to be marginally destabilizing. i.e. E493 becomes slightly more favorable when combined together with KP.3 changes L455S and especially F456L (Figure 8F).
しかし、KP.3 のバックグラウンドでは、逆変異 E493Q はわずかに不安定化しているようでした。つまり、E493 は、KP.3 の L455S の変化、特に F456L と組み合わせると、わずかに有利になります (図 8F)。
Previous studies showed that salt bridge interactions in the BA.2 RBD formed by R493 with E37, E35 and D38 residues in ACE2 are partially lost in the BA.2.86 RBD-ACE2 complex.
以前の研究では、ACE2 の E37、E35、および D38 残基と R493 によって形成される BA.2 RBD の塩橋相互作用は、BA.2.86 RBD-ACE2 複合体では部分的に失われていることが示されました。
In the BA.2.86 RBD-ACE2 complex Q493 forms stable interfacial contacts with D30, K31, N33, H34 and E35 including hydrogen-bonding interaction with the K31 side chain and the carboxyl group of E35 from ACE2.
BA.2.86 RBD-ACE2複合体では、Q493はD30、K31、N33、H34、E35と安定した界面接触を形成し、これにはACE2のK31側鎖およびE35のカルボキシル基との水素結合相互作用が含まれます。
We showed that Q493E can partially rearrange the interactions with more flexible H34, E35 and K31 side-chains.
Q493E は、より柔軟な H34、E35、および K31 側鎖との相互作用を部分的に再配置できることを示しました。
Notably, the salt bridge formed by R493 of BA.2 with E35 of ACE2 is replaced by Q493 in BA.2.75, BF.7, XBB.1 and BA.2.86.
特に、BA.2 の R493 と ACE2 の E35 によって形成される塩橋は、BA.2.75、BF.7、XBB.1、および BA.2.86 では Q493 に置き換えられています。
Although mutational profiling of E493 in the KP.3 ensemble showed that many changes could be stabilizing, the observed changes are small and are often stabilizing for small hydrophobic substituents (Figure 8F).
KP.3 アンサンブルの E493 の変異プロファイリングでは、多くの変化が安定化につながる可能性があることが示されましたが、観察された変化は小さく、多くの場合、小さな疎水性置換基を安定化させるものです (図 8F)。
Overall, the results suggested that position 493 is quite tolerant to modifications and can be mutated in different Omicron variants to balance ACE2 binding and immune evasion.
全体的に、結果は、位置 493 は変更に対して非常に耐性があり、ACE2 結合と免疫回避のバランスをとるために、さまざまな Omicron バリアントで変異できることを示唆しています。
These results are also consistent with the experiments showing that small hydrophobic amino acids for S-Q493 and aromatic amino acids for S-N501 can be highly enriched for ACE2 binding, as these changes are expected to increase local hydrophobic packing.
これらの結果は、S-Q493の小さな疎水性アミノ酸とS-N501の芳香族アミノ酸がACE2結合に非常に豊富であることを示す実験とも一致しており、これらの変化は局所的な疎水性パッキングを増加させると予想される。
This is also in agreement with the DMS experiments.
これはDMS実験とも一致している。
It was conjectured that the fact that these mutations predicted to increase ACE2 binding are not enriched in circulating SARS-CoV-2 variants suggests the affinity of the virus for its receptor is already sufficient for high transmission.
ACE2結合を増加させると予測されるこれらの変異が、循環するSARS-CoV-2変異体では豊富に見られないという事実は、ウイルスの受容体に対する親和性がすでに高い伝染力を発揮するのに十分であることを示唆していると推測された。
Our analysis also confirmed that R493/Q493/E493 in different Omicron variants including latest KP.3 can partially reassemble the interaction network while preserving convergent binding pattern of interactions which may enable evolutionary advantage through epistatic couplings between key hotspots at 456 and 493 positions on the RBD.
私たちの分析では、最新の KP.3 を含むさまざまな Omicron バリアントの R493/Q493/E493 が、RBD 上の 456 と 493 の位置にある主要なホットスポット間のエピスタシス結合を通じて進化上の利点を可能にする可能性のある相互作用の収束結合パターンを維持しながら、相互作用ネットワークを部分的に再構築できることも確認されました。
3.4. MM-GBSA Analysis of the Binding Affinities for the XBB RBD-ACE2 Complexes
XBB RBD-ACE2複合体の結合親和性のMM-GBSA分析
According to the recent experimental studies using accurate SPR measurements the binding affinity of JN.1 RBD-ACE2 complex is KD = 13 nM is reduced as compared to that of BA.2.86 variant (KD = 1.7 nM) which is attributed to a deleterious L455S change in JN.1.
正確なSPR測定を用いた最近の実験研究によると、JN.1 RBD-ACE2複合体の結合親和性はKD = 13 nMで、BA.2.86変異体(KD = 1.7 nM)と比較して低下しており、これはJN.1の有害なL455S変化に起因する。
At the same time, mutations F456L (KD = 12 nM) and R346T + F456L (KD = 11 nM) could not largely affect the ACE2-binding affinity of JN.1 indicating that the dampened ACE2 affinity of JN.1 due to L455S could not be fully compensated by F456L.
同時に、変異 F456L (KD = 12 nM) および R346T + F456L (KD = 11 nM) は、JN.1 の ACE2 結合親和性に大きな影響を与えることができず、L455S による JN.1 の ACE2 親和性の低下が F456L によって完全に補われなかったことを示しています。
The central finding of this illuminating experimental study is that the Q493E mutation of KP.3 can markedly improve the ACE2 binding affinity revealing KD = 6.9 nM which is only marginally lower than the superior binding affinity of the BA.2.86 variant.
この啓発的な実験研究の中心的な発見は、KP.3 の Q493E 変異により ACE2 結合親和性が著しく向上し、KD = 6.9 nM が明らかになったことです。これは、BA.2.86 変異体の優れた結合親和性よりわずかに低いだけです。
Using the conformational equilibrium ensembles obtained fromAF2 predictions and MD simulations we computed the binding free energies for the RBD-ACE2 complexes using the MM-GBSA method.
AF2 予測と MD シミュレーションから得られたコンフォメーション平衡アンサンブルを使用して、MM-GBSA 法を使用して RBD-ACE2 複合体の結合自由エネルギーを計算しました。
To reduce noise and cancel errors in MM-GBSA computations we ran MD simulations on the complexes only, with snapshots taken from a single trajectory to calculate each free energy component.
MM-GBSA 計算におけるノイズを減らし、エラーをキャンセルするために、複合体のみで MD シミュレーションを実行し、単一の軌跡からスナップショットを取得して各自由エネルギー成分を計算しました。
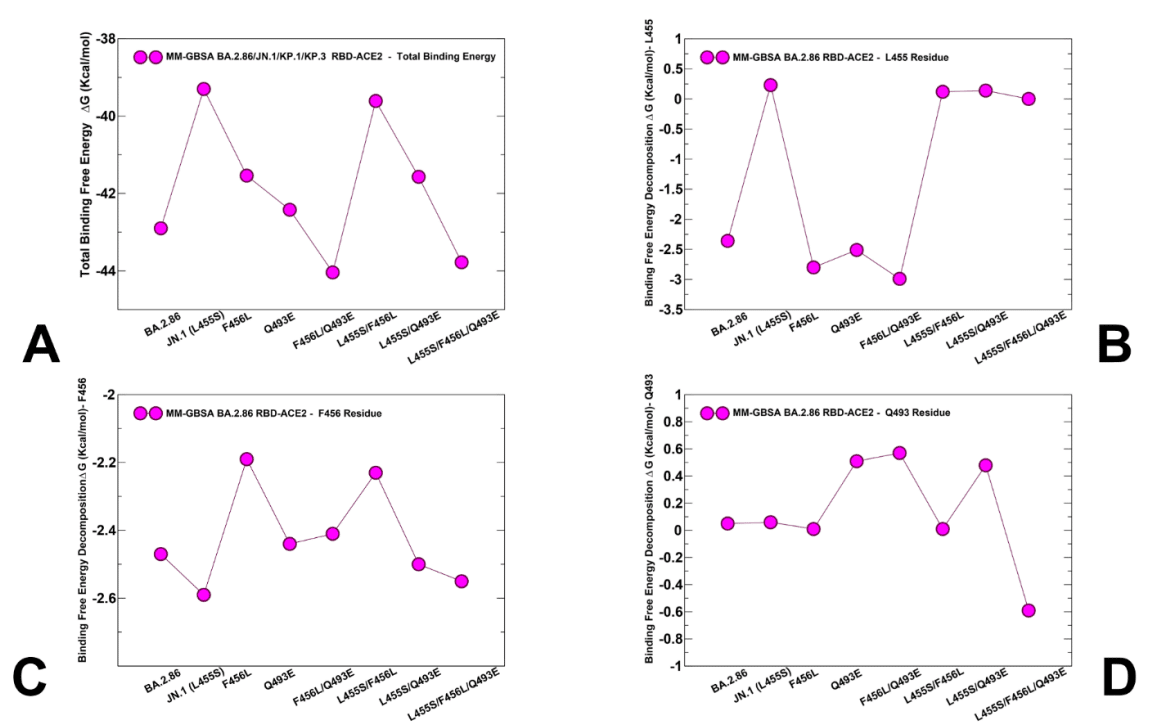
We computed and compared the RBD-ACE2 binding affinities for BA.2.86, JN.1 (BA.2.86+L455S), BA.2.86+F456L, BA.2.86+Q493E, BA.2.86+L455S, BA.2.86+F456L/Q493E, KP.2 (BA.2.86+L455S/F456L/R346T), and KP.3 (BA.2.86+L455S/F456L/Q493E) (Figure 9, Table 2).
BA.2.86、JN.1 (BA.2.86+L455S)、BA.2.86+F456L、BA.2.86+Q493E、BA.2.86+L455S、BA.2.86+F456L/Q493E、KP.2 (BA.2.86+L455S/F456L/R346T)、および KP.3 (BA.2.86+L455S/F456L/Q493E) の RBD-ACE2 結合親和性を計算し、比較しました (図 9、表 2)。
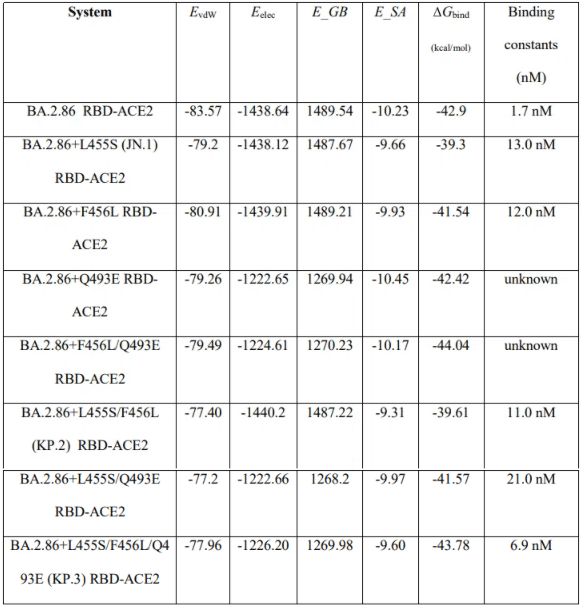
The results of MM-GBSA computations showed a robust agreement with the experimental binding affinities (Figure 9).
MM-GBSA 計算の結果は、実験的な結合親和性との堅実な一致を示しました (図 9)。
The total binding free energy changes showed a more favorable binding affinity for the BA.2.86 RBD-ACE2 with the ΔG = -42.9 kcal/mol (Figure 9A).
結合自由エネルギーの総変化は、ΔG = -42.9 kcal/mol で、BA.2.86 RBD-ACE2 の結合親和性の方が良好であることを示しました (図 9A)。
Consistent with the experiments, we found that L455S in JN.1 can lead to significant reduction in binding with ACE2 as compared to BA.2.86, showing ΔG = -39.3 kcal/mol (Figure 9A).
実験と一致して、JN.1 の L455S は BA.2.86 と比較して ACE2 との結合を大幅に減少させることができ、ΔG = -39.3 kcal/mol を示していることがわかりました (図 9A)。
As may be expected, the breakdown of the MM-GBSA binding energies (Table 2) showed the key role of the favorable van der Waals interactions (ΔG = -83.57 kcal/mol) in BA.2.86 that is markedly more favorable than that in JN.1 (ΔG = -79.20 kcal/mol).
予想どおり、MM-GBSA 結合エネルギーの内訳 (表 2) は、BA.2.86 の好ましいファンデルワールス相互作用 (ΔG = -83.57 kcal/mol) が JN.1 のそれ (ΔG = -79.20 kcal/mol) よりも著しく好ましい重要な役割を担っていることを示しました。
This MM-GBSA analysis confirmed that L455S in JN.1 can reduce the interaction energy and reduce the binding affinity.
この MM-GBSA 分析により、JN.1 の L455S が相互作用エネルギーを減らし、結合親和性を減らすことができることが確認されました。
The energetic analysis of the BA.2.86+F456L complex revealed that F456L alone has only minor effect on the binding affinity resulting in the overall ΔG = -41.54 kcal/mol (Figure 9A,Table 2) which is comparable to that of the BA.2.86 RBD-ACE2 complex.
BA.2.86+F456L 複合体のエネルギー分析により、F456L 単独では結合親和性にわずかな影響しか及ぼさず、全体的な ΔG = -41.54 kcal/mol (図 9A、表 2) となり、BA.2.86 RBD-ACE2 複合体と同等であることが明らかになりました。
These results agree with reported unpublished observations by Starr (https://x.com/tylernstarr/status/1800315116929560965).
これらの結果は、Starr による未発表の観察結果と一致しています。
According to MM-GBSA calculations, the binding affinity of the BA.2.86+Q493E and especially BA.2.86+Q493E/F456L complexes displayed a revealing and interesting trend in which the binding free energy is progressively improved to ΔG = -42.42 kcal/mol and ΔG = - 44.04 kcal/mol (Figure 9, Table 2).
MM-GBSA 計算によると、BA.2.86+Q493E、特に BA.2.86+Q493E/F456L 複合体の結合親和性は、結合自由エネルギーが徐々に ΔG = -42.42 kcal/mol および ΔG = - 44.04 kcal/mol に改善されるという、明らかで興味深い傾向を示しました (図 9、表 2)。
Hence, our predictions indicate that coupling of Q493E and F456L mutations may fully restore binding affinity of the BA.2.86 variant.
したがって、私たちの予測では、Q493E および F456L 変異の結合により、BA.2.86 変異体の結合親和性が完全に回復する可能性があることを示しています。
Interestingly, the MM-GBSA breakdown of the energetic components showed the reduced electrostatic contribution in variants bearing the Q493E mutation, but this reduction is compensated by the decreased polar solvation penalty, leading to the overall more favorable binding energies.
興味深いことに、エネルギー成分の MM-GBSA 分析では、Q493E 変異を持つ変異体では静電寄与が減少していることが示されましたが、この減少は極性溶媒和ペナルティの減少によって補われ、全体として結合エネルギーがより有利になりました。
The comprehensive MM-GBSA analysis is also in line with the experiments by demonstrating that binding affinity of the BA.2.86 L455S/F456L variant (ΔG = -39.61 kcal/mol ) is similar to that of JN.1 (BA.2.86+L455S) and therefore suggesting that F456L mutation alone cannot compensate the loss of binding due to L455S.
包括的な MM-GBSA 分析も実験と一致しており、BA.2.86 L455S/F456L 変異体の結合親和性 (ΔG = -39.61 kcal/mol) は JN.1 (BA.2.86+L455S) の結合親和性と類似していることが示され、したがって F456L 変異だけでは L455S による結合の喪失を補うことができないことが示唆されています。
The important finding of our analysis is that KP.3 variant that bears L455S, F456L and Q493E displayed a considerably improved binding affinity (ΔG = -43.78 kcal/mol) as compared to the individual effects of F456L and Q493E alone (Figure 9A, Table 2).
私たちの分析の重要な発見は、L455S、F456L、および Q493E を持つ KP.3 変異体が、F456L および Q493E 単独の個々の効果と比較して、大幅に改善された結合親和性 (ΔG = -43.78 kcal/mol) を示したことです (図 9A、表 2)。
These results are consistent with the very latest experimental data showing that non-additive epistatic interactions between Q493E and other mutations of KP.3 can represent the energetic driver behind unexpectedly enhanced affinity of KP.3.
これらの結果は、Q493EとKP.3の他の変異との間の非加法的エピスタシス相互作用が、KP.3の予想外に強化された親和性の背後にあるエネルギードライバーとなり得ることを示す最新の実験データと一致しています。
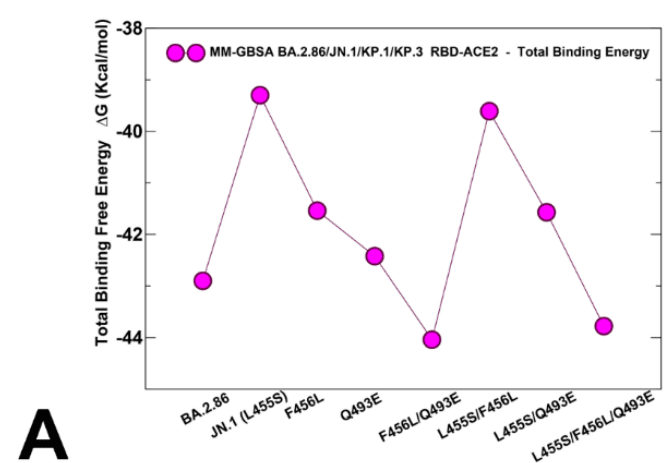
To evaluate the contributions of L455, F456 and Q493 residues and corresponding mutations across all studied variants, we also reported the MM-GBSA residue-based breakdown (Figure 9B,C,D).
研究したすべての変異体における L455、F456、Q493 残基と対応する変異の寄与を評価するために、MM-GBSA 残基ベースの分解も報告しました (図 9B、C、D)。
We found that L455S can induce consistent and similar loss of binding interactions across all variants that share this mutation (Figure 9B), thus indicating that deleterious effect of L455S is not affected by presence of F456L and Q493E mutations.
L455S は、この変異を共有するすべての変異体で一貫して同様の結合相互作用の喪失を引き起こす可能性があることが分かりました (図 9B)。これは、L455S の有害な影響が F456L および Q493E 変異の存在によって影響を受けないことを示しています。
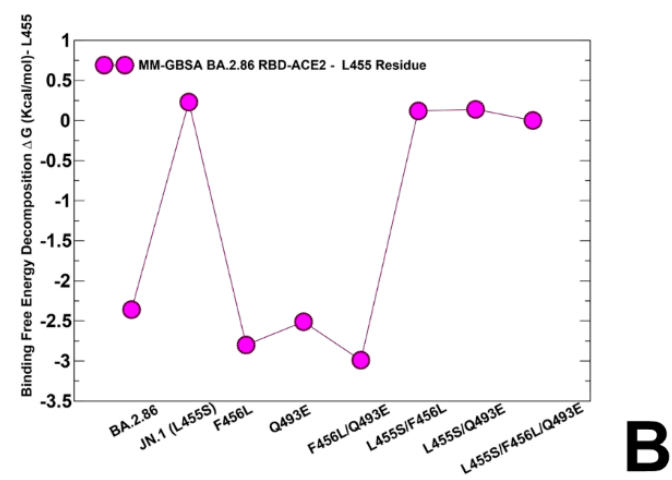
Of particular interest is MM-GBSA decomposition analysis at 456 position (Figure 9).
特に興味深いのは、456 の位置での MM-GBSA 分解分析です (図 9)。
We noticed that F456L can induce loss of binding interactions in the BA.2.86+F456L, while partly reversing the trend and restoring more favorable contribution in BA.2.86+F456L/Q493E and KP.3 (BA.2.86+L455S/F456L/Q493E) variants (Figure 9C).
F456L は BA.2.86+F456L で結合相互作用の喪失を引き起こす一方で、BA.2.86+F456L/Q493E および KP.3 (BA.2.86+L455S/F456L/Q493E) バリアントでは部分的に傾向を逆転させ、より好ましい寄与を回復できることに気付きました (図 9C)。
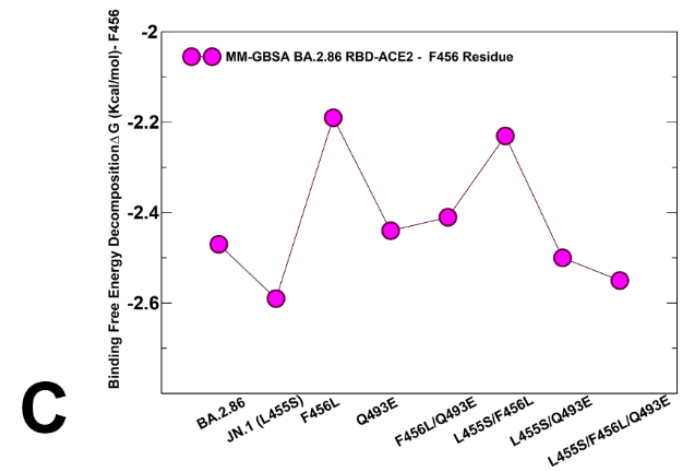
An overall similar trend is seen in the MM-GBSA decomposition of Q493 position (Figure 9D) where small improvements in the contribution of 493 position could be observed in the KP.3 variant.
全体的に同様の傾向が Q493 位置の MM-GBSA 分解で見られ (図 9D)、KP.3 バリアントでは 493 位置の寄与のわずかな改善が見られました。
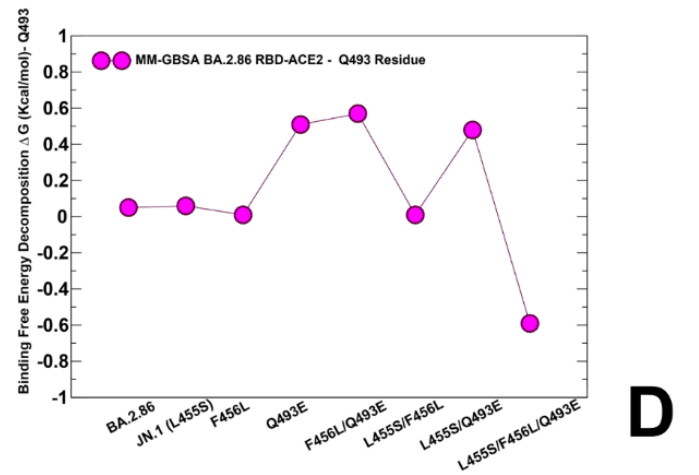
However, the differences between contributions of Q493/E493 in different variants are relatively minor.
しかし、異なる変異体における Q493/E493 の寄与の違いは比較的小さい。
We also evaluated contributions of individual ACE2 positions H34 and K31 that are involved in interactions with L455, F456 and Q493 sites (Supporting Information, Figure S5).
また、L455、F456、Q493 部位との相互作用に関与する個々の ACE2 位置 H34 および K31 の寄与も評価した (補足情報、図 S5)。
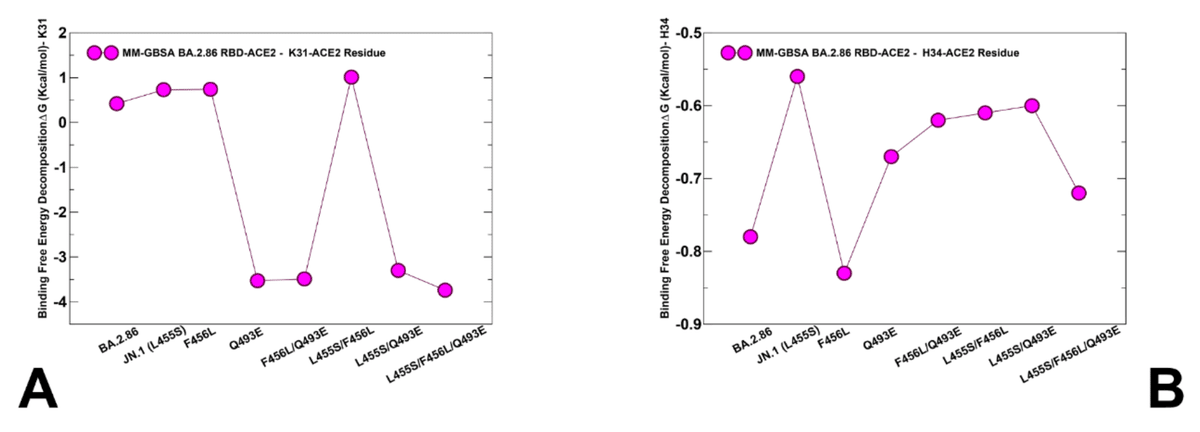
It can be seen that that BA.2.86 variants with F456L/Q493E and L455S/F456L/Q493E additions lead to stronger contribution of K31 position on ACE2 due to favorable combination of van der Waals and electrostatic interactions (Supporting Information, Figure S4A).
F456L/Q493E および L455S/F456L/Q493E を追加した BA.2.86 バリアントは、ファンデルワールス相互作用と静電相互作用の好ましい組み合わせにより、ACE2 上の K31 位置の寄与が強くなることがわかります (補足情報、図 S4A)。
In BA.2.86 Q493 is hydrogen-bonded with K31 while in F456L/Q493E and L455S/F456L/Q493E variants, E493 and K31 form strong ionic electrostatic interactions and there are more van der Waals contacts between Q493E and K31 of ACE2 (Supporting Information, Figure S5A).
BA.2.86 では Q493 は K31 と水素結合していますが、F456L/Q493E および L455S/F456L/Q493E バリアントでは、E493 と K31 が強力なイオン静電相互作用を形成し、ACE2 の Q493E と K31 の間にはより多くのファンデルワールス接触があります (補足情報、図 S5A)。
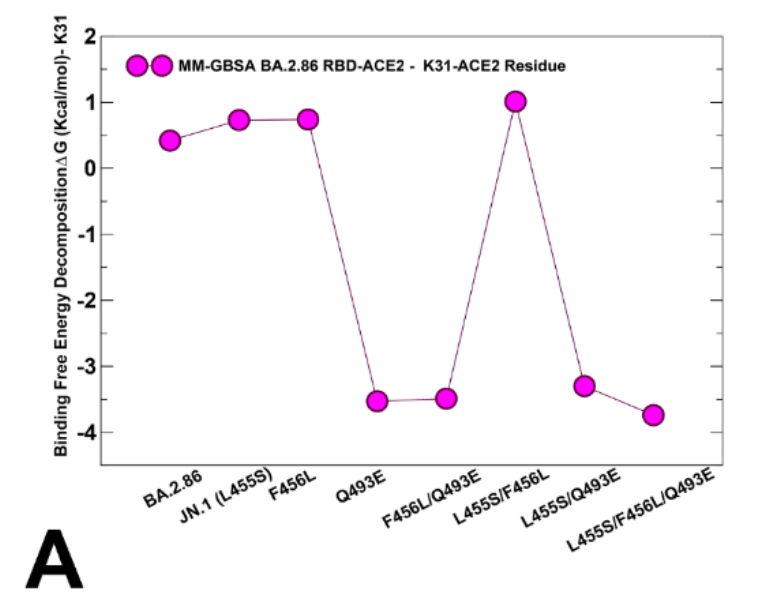
We also found that contribution of H34 becomes more favorable in variants sharing F456L ((Supporting Information, Figure S5B).
また、F456Lを共有する変異体では、H34の寄与がより有利になることもわかりました(補足情報、図S5B)。
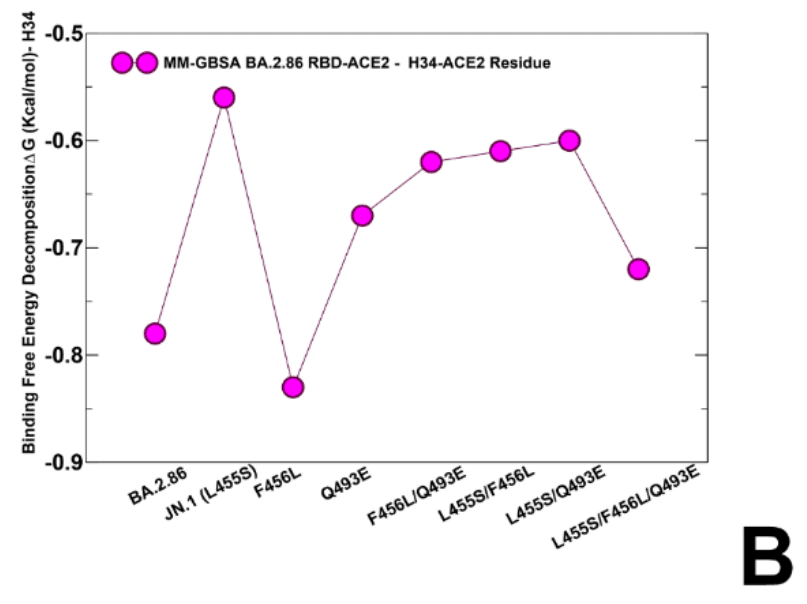
Moreover, multiple side-chain conformations of H34 are involved in multiple contacts and form favorable interactions with Y453.
さらに、H34の複数の側鎖コンフォメーションは複数の接触に関与し、Y453との好ましい相互作用を形成します。
Together, structural and energetic analysis of BA.2.86 combinations including JN.1, KP.2 and KP.3 variants suggested that F456L and Q493E may act cooperatively in KP.3 to induce epistatic couplings and restore the binding affinity to the level of BA.2.86.
JN.1、KP.2、KP.3変異体を含むBA.2.86の組み合わせの構造およびエネルギー分析を合わせると、F456LとQ493EがKP.3で協力してエピスタシス結合を誘導し、結合親和性をBA.2.86のレベルに回復させる可能性があることが示唆されました。
Although precise molecular mechanisms underlying the observed epistatic effects of Q493E and F456L are likely to be complex, our results suggested that the increased flexibility of the RBD in the KP.3 complex may allow for more plasticity of the reciprocal ACE2 sites H34 and K31 that appeared to adopt multiple side-chain conformations in the structural ensembles.
Q493EとF456Lの観察されたエピスタシス効果の背後にある正確な分子メカニズムは複雑である可能性が高いが、我々の結果は、KP.3複合体のRBDの柔軟性の向上により、構造アンサンブルで複数の側鎖コンフォメーションを採用しているように見える相互ACE2部位H34とK31の可塑性が高まる可能性があることを示唆した。
We found the side chain of H34 adopts two alternative conformations that are also seen in the BA.4/5 complex and forms hydrogen bonding with Y453.
H34 の側鎖は、BA.4/5 複合体でも見られる 2 つの代替コンフォメーションを採用し、Y453 と水素結合を形成することを発見した。
Structural examination of the predicted ensembles and MM-GBSA analysis of the RBD-ACE2 complexes for JN.1, KP.2 and KP.3 variants suggested that the plasticity of the ACE2 interfacial sites that are afforded by L455S/F456L mutations can be properly exploited by Q493E, Y453 and F456L positions to enhance binding affinity.
JN.1、KP.2、KP.3 変異体の RBD-ACE2 複合体の予測アンサンブルの構造検査と MM-GBSA 分析により、L455S/F456L 変異によってもたらされる ACE2 界面部位の可塑性は、Q493E、Y453、F456L の位置によって適切に利用され、結合親和性を高めることができることが示唆されました。
Overall, this analysis pointed to a central role of F456L as a potential regulator of epistatic couplings but also suggests a potential role of structurally proximal L452 and Y453 sites supporting the improved binding interfacial interactions in the KP.3 variant.
全体として、この分析は、エピスタシス結合の潜在的な調節因子としての F456L の中心的な役割を指摘しましたが、KP.3 変異体における結合界面相互作用の改善をサポートする構造的に近接した L452 および Y453 部位の潜在的な役割も示唆しています。
3.5. Mutational Profiling of Protein Binding Interfaces with Distinct Classes of Antibodies
異なるクラスの抗体によるタンパク質結合界面の変異プロファイリング
We embarked on structure-based mutational analysis of the S protein binding with different classes of RBD-targeted antibodies, focusing specifically on the role of BA.2.86 mutations in mediating potential resistance to broad class of antibodies and eliciting robust immune escape.
我々は、RBDを標的としたさまざまなクラスの抗体と結合するSタンパク質の構造に基づく変異解析に着手し、特にBA.2.86変異が幅広いクラスの抗体に対する潜在的な耐性を媒介し、強力な免疫回避を誘発する役割に焦点を当てました。
We specifically examined a panel of monoclonal antibodies that were reported to retain activity against BA.2. XBB.1.5 and BA.2.86 variant but showed reduced neutralization against JN.1, KP.2 and KP.3 variants.
我々は特に、BA.2.XBB.1.5およびBA.2.86変異体に対して活性を保持するが、JN.1、KP.2およびKP.3変異体に対しては中和効果が低下すると報告されているモノクローナル抗体のパネルを調べた。
Structure-based mutational scanning of the S protein binding interfaces with a panel of class I RBD-targeting antibodies included S2K146, Omi-3 Omi-18, Omi-42 as well as antibodies BD55-5514 (SA55) and BD55-5840 (SA58) that bind to a different RBD epitope and are experimentally known to tolerate escape mutations in BA.2.86, JN.1, KP.2 and KP.3 variants.
Sタンパク質結合インターフェースの構造に基づく変異スキャンには、クラスI RBD標的抗体のパネルが含まれており、S2K146、Omi-3 Omi-18、Omi-42、および抗体BD55-5514(SA55)とBD55-5840(SA58)が含まれます。 これらは異なるRBDエピトープに結合し、実験的にBA.2.86、JN.1、KP.2、およびKP.3バリアントの回避変異を許容することが知られています。
The results were compared with biochemical measurements from a panel of broadly neutralizing monoclonal antibodies.
結果は、広範囲に中和するモノクローナル抗体のパネルからの生化学的測定値と比較されました。
In particular, the experiments found that L455S of JN.1 significantly enhances immune evasion at the expense of reduced ACE2 binding affinity.
特に、実験では、JN.1のL455SがACE2結合親和性の低下を犠牲にして免疫回避を大幅に強化することが判明した。
To provide a systematic comparison, we constructed mutational heatmaps for the RBD interface residues of the S complexes with S2K146 (Figure 10A), OMI-3 (Figure 10B), OMI-18 (Figure 10C) and OMI-42 antibodies (Figure 10D).
体系的な比較を行うために、S2K146(図10A)、OMI-3(図10B)、OMI-18(図10C)、およびOMI-42抗体(図10D)を含むS複合体のRBDインターフェース残基の変異ヒートマップを作成しました。
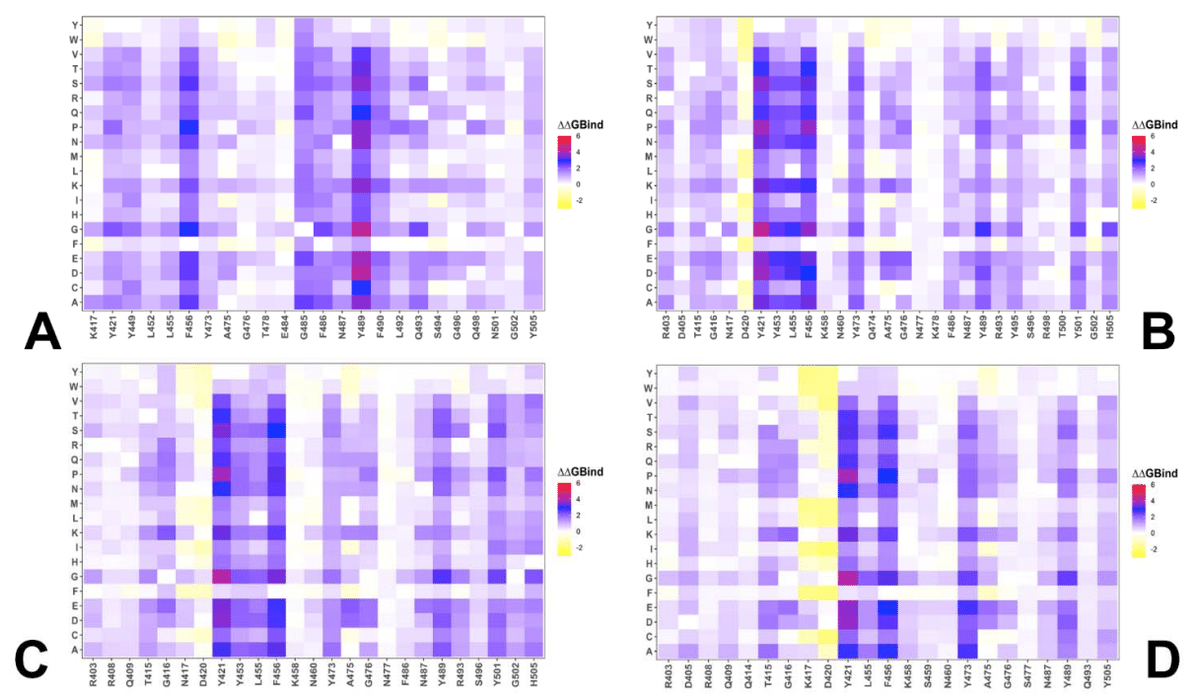
Strikingly, in all S complexes with class I antibodies mutational heatmaps clearly showed that a pair of adjacent residues L455 and F456 corresponding to convergent evolutionary hotspots are also dominant escape hotpots of antibody neutralization.
驚くべきことに、クラスI抗体を含むすべてのS複合体において、変異ヒートマップは、収束進化ホットスポットに対応する隣接する残基L455とF456のペアが、抗体中和の主要な回避ホットスポットでもあることを明らかに示しました。
These results are consistent with the experimental data showing that antibody evasion drives the convergent evolution of L455F/S and F456L, while the epistatic shift caused by F456L can facilitate the subsequent convergence of L455 and Q493 changes to restore ACE2 binding.
これらの結果は、抗体の回避がL455F/SとF456Lの収束進化を駆動し、F456Lによって引き起こされるエピスタシスシフトがその後のL455とQ493の変化の収束を促進してACE2結合を回復することを示す実験データと一致しています。
Furthermore, mutational heatmaps indicated that other important positions involved in JN.1, KP.2 and KP.3 variants, particularly Q493 site can be considered as a secondary escape hotspot sine mutations of Q493 across all class I antibodies (Figure 10).
さらに、変異ヒートマップは、JN.1、KP.2、および KP.3 バリアントに関与する他の重要な位置、特に Q493 部位が、すべてのクラス I 抗体における Q493 の変異による二次エスケープ ホットスポットとして考えられることを示しています (図 10)。
Interestingly, most of the RBD residues that directly bind S2K146 are also involved in binding to ACE2.
興味深いことに、S2K146 に直接結合する RBD 残基のほとんどは、ACE2 への結合にも関与しています。
Indeed, the mutational heatmap of the RBD binding against S2K146 identified that all substitutions in key interfacial positions can incur a consistent and considerable loss in binding affinity with S2K146 (Figure 10A).
実際、S2K146 に対する RBD 結合の変異ヒートマップでは、主要な界面位置のすべての置換が、S2K146 との結合親和性の一貫した大幅な低下を引き起こす可能性があることが特定されました (図 10A)。
Among these sites are F456, F486, N487 , Y489, F490, and Q493 as mutations in these sites caused the largest losses in the binding affinity with S2K146 (Figure 10A ).
これらの部位には、F456、F486、N487、Y489、F490、Q493があり、これらの部位の変異により、S2K146との結合親和性が最も低下しました(図10A)。
These findings are full consistent structural studies showing that the S2K146 footprint on the SARS-CoV-2 RBD mimics that of the ACE2 receptor, with 18 of 24 epitope residues shared with the ACE2 binding site, including L455, F486, Q493, Q498, and N501.
これらの知見は、SARS-CoV-2 RBD上のS2K146フットプリントがACE2受容体のフットプリントを模倣していることを示す、完全に一貫した構造研究です。 24個のエピトープ残基のうち18個がACE2結合部位と共有されており、これにはL455、F486、Q493、Q498、N501が含まれます。
The important finding of the mutational heatmap analysis is that positions Y421, Y453, L455 and F456 emerged as key escape hotspots in S binding with Omi-3, Omi-18 and Omi-42 class I antibodies (Figure 10B-D).
変異ヒートマップ解析の重要な発見は、Y421、Y453、L455、F456の位置が、Omi-3、Omi-18、Omi-42クラスI抗体とのS結合における主要なエスケープホットスポットとして現れたことです(図10B-D)。
L455 and F456 positions are located at the epitope of RBD Class 1 antibodies and neutralization assays demonstrated that L455S mutation enables JN.1 to evade Class 1 antibodies.
L455とF456の位置はRBDクラス1抗体のエピトープに位置しており、中和アッセイでは、L455S変異によりJN.1がクラス1抗体を回避できることが実証されました。
Mutational heatmaps data are consistent with these experiments, showing that effectively all modifications in L455, including L455S, can cause considerable loss in antibody binding.
変異ヒートマップデータはこれらの実験と一致しており、L455Sを含むL455のすべての変更が抗体結合の大幅な低下を引き起こす可能性があることを示しています。
In addition, the results also highlighted the importance of Y453 position that is involved in favorable interactions with the ACE2 and antibodies.
さらに、結果は、ACE2および抗体との好ましい相互作用に関与するY453の位置の重要性も強調しました。
A secondary group of escape hotspots for these class I antibodies included F486, N487, Y489, and Q493 positions (Figure 10B-D).
これらのクラスI抗体のエスケープホットスポットの2番目のグループには、F486、N487、Y489、およびQ493の位置が含まれます(図10B-D)。
Mutations in some of these sites such as F486P are implicated as the main immune escape hotspots for BA.2.86 variant.
F486Pなどのこれらの部位の一部の変異は、BA.2.86変異体の主な免疫エスケープホットスポットとして関与しています。
Finally, due to mimicry of the ACE2 binding, other escape positions correspond to the ACE2 binding affinity hotspots Y501 and H505 (Figure 10).
最後に、ACE2 結合の模倣により、他のエスケープ位置は ACE2 結合親和性ホットスポット Y501 および H505 に対応します (図 10)。
Importantly, the result firmly suggested that major drivers of immune escape for this class I antibodies correspond to L455 and F456 sites that undergo mutations in the JN.1, KP.2 and KP.3 variants, enabling evolution through the enhanced immune escape.
重要なことに、この結果は、このクラス I 抗体の免疫エスケープの主な要因が、JN.1、KP.2、および KP.3 バリアントで変異を受ける L455 および F456 サイトに対応することを確実に示唆しています。 免疫エスケープの強化による進化を可能にします。
We then examined JN.1, KP.2 and KP.3 mutations using BA.286 as genetic background to quantify binding free energy changes of S binding with the class I antibodies (Figure 11).
次に、BA.286 を遺伝的背景として JN.1、KP.2、および KP.3 変異を調べ、クラス I 抗体との S 結合の結合自由エネルギー変化を定量化しました (図 11)。
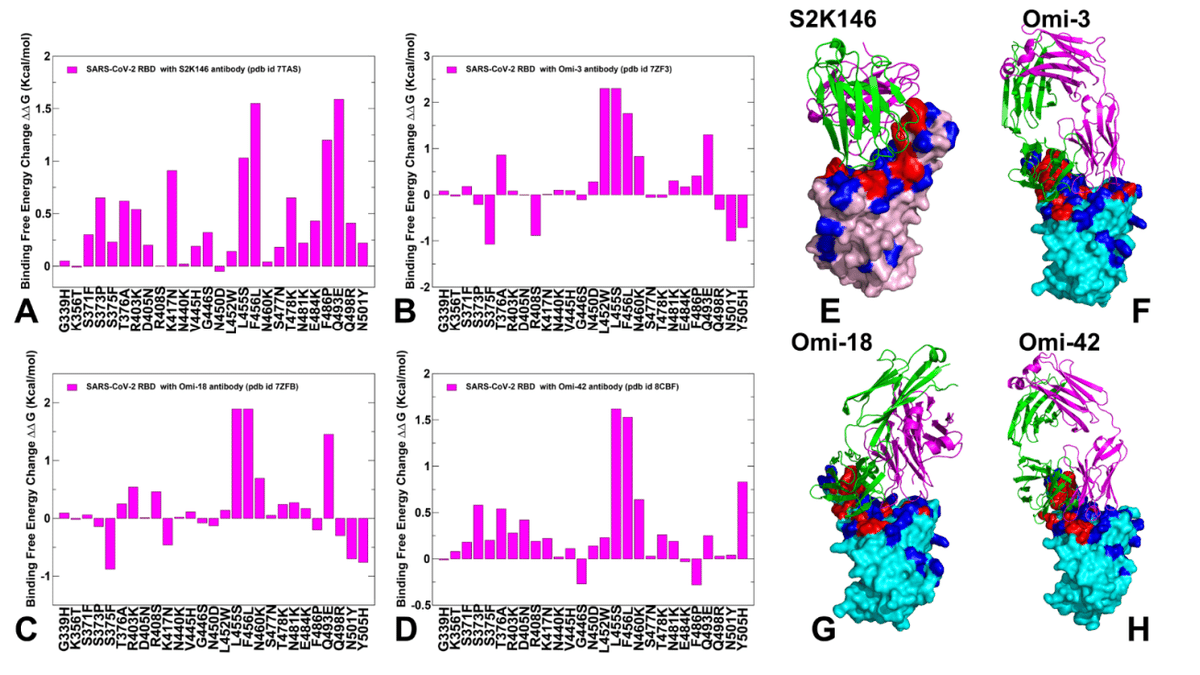
The binding free energy changes associated with BA.2.86 mutations in the complex with S2K146 (Figure 11A) showed an appreciable loss of binding upon K417N, L455, F456L, F486P, Q493E and Q498R mutations.
S2K146 との複合体における BA.2.86 変異に関連する結合自由エネルギー変化 (図 11A) は、K417N、L455、F456L、F486P、Q493E、および Q498R 変異による結合の顕著な損失を示しました。
Interestingly, the largest destabilization changes were induced L455S ( ΔΔG = 1.03 kcal/mol), F456L (ΔΔG = 1.55 kcal/mol) and Q493E (ΔΔG = 1.59kcal/mol).
興味深いことに、最も大きな不安定化変化は、L455S (ΔΔG = 1.03 kcal/mol)、F456L (ΔΔG = 1.55 kcal/mol)、および Q493E (ΔΔG = 1.59kcal/mol) によって誘発されました。
Hence, these key mutational changes present in JN.1, KP.2 and KP.3 variants may induce progressively enhanced immune escape from S2K146 which is consistent with the experiments.
したがって、JN.1、KP.2、および KP.3 変異体に存在するこれらの重要な変異は、S2K146 からの免疫逃避を徐々に強化する可能性があり、これは実験と一致しています。
We found that L455S and F456L mutations induce very significant losses in antibody binding with Omi-3, Omi-18 and Omi-42 antibodies (Figure 11B-D).
L455S および F456L 変異は、Omi-3、Omi-18、および Omi-42 抗体との抗体結合において非常に大きな損失を引き起こすことがわかりました (図 11B-D)。
In particular, for binding with Omi-3, L455S mutation caused ΔΔG = 2.3 kcal/mol and F456L incurred ΔΔG = 1.76 kcal/mol (Figure 11B), while for Omi-42 these losses were ΔΔG = 1.62 kcal/mol for L455S and ΔΔG = 1.53 kcal/mol for F456 (Figure 11D).
特に、Omi-3 との結合では、L455S 変異により ΔΔG = 2.3 kcal/mol が発生し、F456L により ΔΔG = 1.76 kcal/mol が発生しました (図 11B)。一方、Omi-42 の場合、これらの損失は L455S で ΔΔG = 1.62 kcal/mol、F456 で ΔΔG = 1.53 kcal/mol でした (図 11D)。
These results are consistent with the experiments showing the increased evasion against JN.1 variant with Omi-3 and Omi-18 antibodies as L455 and F456 sites emerge as dominant hotspots where mutations L455S and F456L caused the largest loss in affinity.
これらの結果は、Omi-3 および Omi-18 抗体による JN.1 変異体に対する回避の増加を示す実験と一致しており、L455 および F456 サイトが主要なホットスポットとして現れ、変異 L455S および F456L が最大の親和性の低下を引き起こしました。
Common to both S2K146 and Omi-3 antibodies, we also observed a considerable loss of binding due to F486P mutation (Figure 11A,B).
S2K146 および Omi-3 抗体の両方に共通して、F486P 変異による結合の大幅な低下も観察されました (図 11A、B)。
Specific for Omi-3 antibody is a destabilizing role of N460K mutation inducing loss of binding ΔΔG = 0.83 kcal/mol (Figure 11B).
Omi-3 抗体に特有ののは、結合の低下 ΔΔG = 0.83 kcal/mol を引き起こす N460K 変異の不安定化の役割です (図 11B)。
Interestingly, Q493E emerges as the third most important escape mutation in binding with Omi-3 and Omi-18 antibodies (Figure 11B,C).
興味深いことに、Q493E は Omi-3 および Omi-18 抗体との結合において 3 番目に重要なエスケープ変異として現れています (図 11B、C)。
Structural mapping of the binding epitope residues and sites of BA.2.86 mutations (Figure 11E-H) highlighted similar binding modes for these antibodies, targeting the second region of the RBD that includes R403, L455, Y453, and Q493 sites.
BA.2.86 変異の結合エピトープ残基と部位の構造マッピング (図 11E-H) により、これらの抗体の結合モードが類似していることが強調されました。 R403、L455、Y453、および Q493 部位を含む RBD の 2 番目の領域をターゲットにします。
In general, the results confirmed the experimental finding that KP.2 and KP.3 harboring the F456L mutation, alone or in combination with Q493E (KP.3) can significantly impair the neutralizing activity of RBD class-1 monoclonal antibodies such as BD-1854, BD57- 1302, and Omi-42.
一般的に、結果は、F456L変異を有するKP.2およびKP.3が、単独またはQ493E(KP.3)との組み合わせで、BD-1854、BD57-1302、およびOmi-42などのRBDクラス1モノクローナル抗体の中和活性を著しく損なう可能性があるという実験的発見を裏付けました。
BD55-5514 belongs to class F2 and F3 antibodies that compete with ACE2, and their binding is affected by T376, K378, D405, R408 and G504.
BD55-5514はACE2と競合するクラスF2およびF3抗体に属し、その結合はT376、K378、D405、R408、およびG504の影響を受ける。
BD55-5514 displayed high potency against the Omicron subvariants and recently developed non-competing antibody cocktail of BD55-5840 (also known as SA58; class 3) and BD55-5514 (also known as SA55; class 1/4), displayed high potency against the Omicron subvariants.
BD55-5514 はオミクロン亜種に対して高い効力を示し、最近開発された BD55-5840 (別名 SA58、クラス 3) と BD55-5514 (別名 SA55、クラス 1/4) の非競合抗体カクテルはオミクロン亜種に対して高い効力を示した。
Recent studies by Cao group showed that BD55-5514 (SA55) antibody can retain neutralizing efficacy against most of the known Omicron variants including JN.1.
Cao グループの最近の研究では、BD55-5514 (SA55) 抗体は、JN.1 を含む既知の Omicron 変異体のほとんどに対して中和効果を維持できることが示されています。
SA55 antibody also showed significant neutralization activity against other variants sharing F456L mutation including HV.1 (L452R+F456L), JD.1.1(L455F/F456L+A475V) that typically greatly increase antibody evasion for other class I antibodies at the cost of ACE2 binding.
SA55 抗体は、ACE2 結合を犠牲にして他のクラス I 抗体の抗体回避を大幅に増加させることが多い HV.1 (L452R+F456L)、JD.1.1 (L455F/F456L+A475V) などの F456L 変異を共有する他の変異体に対しても、顕著な中和活性を示しました。
The detailed mutational heatmap of the BD55-5514 interactions with the S protein showed a very different picture as compared to class I antibodies such as Omi-3, Omi-18 and Omi-42 (Figure 12A).
BD55-5514 と S タンパク質の相互作用の詳細な変異ヒートマップは、Omi-3、Omi-18、Omi-42 などのクラス I 抗体と比較して非常に異なる図を示しました (図 12A)。
The map showed that the S373P and S375F mutations could promote the interaction with BD55-5514.
マップは、S373P および S375F 変異が BD55-5514 との相互作用を促進できることを示しました。
T376, D405, and R408 are involved in the interaction with BD55-5514 but they are all located at the periphery of the BD55-5514 epitope, and mutations in these positions have moderate effect on binding affinity (Figure 12A).
T376、D405、および R408 は BD55-5514 との相互作用に関与していますが、これらはすべて BD55-5514 エピトープの周辺に位置しており、これらの位置での変異は結合親和性に中程度の影響を及ぼします (図 12A)。
Importantly, BD55-5514 binding interface does not involve escaping hotspot positions of BA.2.86, JN.1 and KP.2/KP.3 such as L455, F456 and Q493 (Figure 12A).
重要なのは、BD55-5514 結合インターフェースは、L455、F456、Q493 などの BA.2.86、JN.1、KP.2/KP.3 のホットスポット位置を回避しないことです (図 12A)。
A more detailed profiling of JN.1/KP.3 mutations against BD55-5514 antibody showed only small destabilization changes upon mutations T376A ( ΔΔG = 0.81 kcal/mol), R403K ( ΔΔG = 0.65 kcal/mol), D405N (ΔΔG = 0.79 kcal/mol), R408S (ΔΔG = 0.34 kcal/mol), L455S ( ΔΔG = 0.7 kcal/mol) and F456L (ΔΔG = 0.51 kcal/mol) (Figure 12B).
BD55-5514 抗体に対する JN.1/KP.3 変異のより詳細なプロファイリングでは、変異 T376A (ΔΔG = 0.81 kcal/mol)、R403K (ΔΔG = 0.65 kcal/mol)、D405N (ΔΔG = 0.79 kcal/mol)、R408S (ΔΔG = 0.34 kcal/mol)、L455S (ΔΔG = 0.7 kcal/mol)、および F456L (ΔΔG = 0.51 kcal/mol) によるわずかな不安定化の変化のみが示されました (図 12B)。
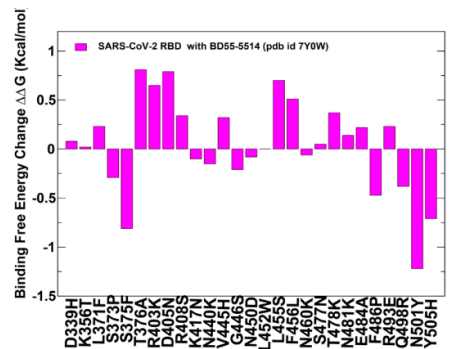
These changes reflect mostly a mild loss in the RBD stability and binding interactions, which is consistent with functional experiments showing group F3 antibodies, such as BD55-5514, are not sensitive to the D405N and R408S mutations of BA.2 making this antibody effective against a broad spectrum of recent variants from BA.2.86 to KP.2 and KP.3.
これらの変化は主にRBDの安定性と結合相互作用の軽度の低下を反映しており、これはBD55-5514などのグループF3抗体がBA.2のD405NおよびR408S変異に感受性がなく、この抗体がBA.2.86からKP.2およびKP.3までの最近の変異体の広範囲に対して有効であることを示す機能実験と一致しています。
Structural mapping of the binding epitopes and BA.2.86 mutational sites for BD55-5514 and BD55-5840 antibodies (Figure 12 C,D) illustrated how BD55-5514 can bind to the RBD together with BD55-5840 without interference.
BD55-5514 および BD55-5840 抗体の結合エピトープと BA.2.86 変異部位の構造マッピング (図 12 C、D) は、BD55-5514 が BD55-5840 とともに干渉なく RBD に結合する様子を示しています。
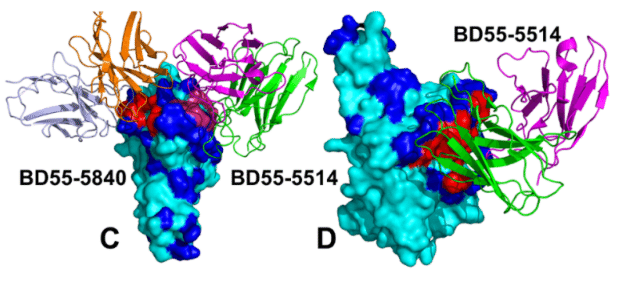
Mutational analysis of the BD55-5514 and BD55-5840 interactions confirmed that S373P and S375F mutations can improve the binding affinities of these antibodies with the S protein which is also consistent with the pseudo-virus data.
BD55-5514 と BD55-5840 の相互作用の変異解析により、S373P および S375F 変異がこれらの抗体と S タンパク質の結合親和性を改善できることが確認されました。これは擬似ウイルスデータとも一致しています。
We also found that binding of BD55-5840 antibody is even less affected by mutations T376A, R403K and D405N as compared to BD55- 5514 (Figure 12E).
また、BD55-5840 抗体の結合は、BD55-5514 と比較して、変異 T376A、R403K、および D405N による影響がさらに少ないこともわかりました (図 12E)。
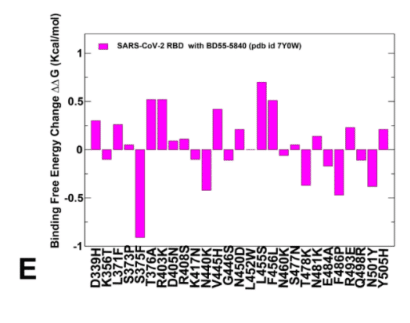
Importantly, both L455S and F456L mutations are only marginally deficient for binding of these antibodies with ΔΔG ~0.5-0.7 kcal/mol (Figure 12A,E).
重要なのは、L455S および F456L 変異はいずれも、ΔΔG が約 0.5~0.7 kcal/mol で、これらの抗体の結合にわずかに欠けているだけであることです (図 12A、E)。
As a results, these antibodies could potently bind to the entire spectrum of BA.2.86, JN.1, KP.2 and KP.3 mutations.
その結果、これらの抗体は、BA.2.86、JN.1、KP.2、および KP.3 変異の全スペクトルに強力に結合することができました。
Our data enabled the quantitative energetic analysis of the binding interactions for these antibodies in the complex where BD55-5840 and BD55-5514 act synergistically and bind to different sides of the RBD (Figure 12C).
我々のデータにより、BD55-5840 と BD55-5514 が相乗的に作用し、RBD の異なる側に結合する複合体におけるこれらの抗体の結合相互作用の定量的なエネルギー分析が可能になりました (図 12C)。
Interestingly, structural mapping of the BA.2.86 mutational sites onto the RBD complex with BD55-5514/BD55-5840 highlighted that binding modes of antibodies do not significantly overlap with the mutational positions (Figure 12 C) and, as a result, this antibody combination can exhibit exceptional neutralizing activity by successfully avoiding immune escape hotspots.
興味深いことに、BD55-5514/BD55-5840 を含む RBD 複合体への BA.2.86 変異部位の構造マッピングにより、抗体の結合モードが変異位置と大幅に重複していないことが明らかになりました (図 12 C)。その結果、この抗体の組み合わせは、免疫逃避ホットスポットをうまく回避することで、並外れた中和活性を発揮できます。
Together, structural and energetic analysis provides a rationale to the experimental results showing that BD55-5840 (SA58) + BD55-5514 (SA55) cocktail exhibits remarkable neutralization breadth and potency and particularly SA55 can retain neutralizing efficacy against all examined variants BA.2.86, JN.1, KP.2 and KP.3.
構造解析とエネルギー解析を組み合わせることで、BD55-5840(SA58)+ BD55-5514(SA55)カクテルが顕著な中和幅と効力を示し、特にSA55は検査したすべての変異体BA.2.86、JN.1、KP.2、KP.3に対して中和効果を維持できることを示す実験結果の根拠が得られます。
A very recent study introduced CYFN1006-1 and CYFN1006-2 antibodies that demonstrated consistent neutralization of all tested SARS-CoV-2 variants comparable to or even superior to those of SA55.
ごく最近の研究では、CYFN1006-1 および CYFN1006-2 抗体が発表され、試験されたすべての SARS-CoV-2 変異体に対して、SA55 と同等かそれ以上の一貫した中和効果を示した。
CYFN1006-2 exhibited high potency against all SARS-CoV-2 variants, with a slightly reduced efficacy against KP.2.
CYFN1006-2は、すべてのSARS-CoV-2変異体に対して高い効力を示したが、KP.2に対する効力はわずかに低下した。
These antibodies have binding epitopes that overlap with LY-CoV1404, REGN10987 and S309 that are situated on the outer surface of RBD and bind to a different RBD region compared to SA55.
これらの抗体は、RBDの外表面に位置し、SA55とは異なるRBD領域に結合するLY-CoV1404、REGN10987、S309と重複する結合エピトープを持っています。
Hence, a cocktail with combinations of SA55 and CYFN1006-1 may be potentially beneficial against JN.1, KP.2, KP.3 and evolving mutants of SARS-CoV-2.
したがって、SA55とCYFN1006-1を組み合わせたカクテルは、JN.1、KP.2、KP.3、およびSARS-CoV-2の進化する変異体に対して潜在的に有益である可能性があります。
4. Discussion
The results of this study provided molecular rationale and support to the experimental evidence that functionally balanced substitutions that optimize tradeoffs between immune evasion, high ACE2 affinity and sufficient conformational adaptability might be a common strategy of the virus evolution and serve as a primary driving force behind the emergence of new Omicron subvariants, including but not limited to BA.2.86, JN.1, KP.2 and KP.3 variants.
この研究の結果は、免疫回避、高い ACE2 親和性、十分な立体構造適応性の間のトレードオフを最適化する機能的にバランスのとれた置換がウイルス進化の一般的な戦略であり、BA.2.86、JN.1、KP.2、KP.3 バリアントを含むがこれらに限定されない新しい Omicron サブバリアントの出現の主な原動力となっている可能性があるという実験的証拠に対する分子論的根拠と裏付けを提供しました。
Recent studies of emerging Omicron variants, particularly evolving BA.2.86 sublineage suggested that the evolutionary paths for significant improvements in the binding affinity of the Omicron RBD variants with ACE2 are relatively narrow and may involve convergent mutational hotspots to primarily optimize immune escape while retaining sufficient ACE2 affinity.
出現しつつあるオミクロン変異体、特に進化中の BA.2.86 亜系統に関する最近の研究では、オミクロン RBD 変異体と ACE2 の結合親和性を大幅に改善する進化の道筋は比較的狭く、十分な ACE2 親和性を維持しながら免疫回避を主に最適化するための収束的変異ホットスポットが関与している可能性があることが示唆されました。
These mechanisms based on convergent adaptation may determine the scope of “ evolutionary opportunities” for the virus to adapt new mutations that improve immune resistance without compromising ACE2 binding affinity and stability.
収束適応に基づくこれらのメカニズムは、ACE2 結合親和性と安定性を損なうことなく免疫抵抗性を改善する新しい変異をウイルスが適応させるための「進化の機会」の範囲を決定する可能性があります。
Our multifaceted study explored quantitative aspects of structure, dynamics and energetics of the BA.2.86, JN.1 and KP.2/KP.3 variants, showing a robust agreement with a large body of experimental data on ACE2 binding and interactions with various classes of antibodies.
私たちの多面的な研究では、BA.2.86、JN.1、KP.2/KP.3 変異体の構造、ダイナミクス、エネルギーの定量的側面を調査し、ACE2 結合およびさまざまなクラスの抗体との相互作用に関する大量の実験データと確実に一致していることを示しました。
The results of our investigation suggested that the existence of epistatic interactions between convergent mutational sites at L455, F456, Q493 positions that enable to protect and restore ACE2 binding affinity while conferring beneficial immune escape.
調査の結果、L455、F456、Q493 の位置にある収束変異部位間のエピスタシス相互作用の存在が示唆されました。この相互作用により、ACE2 結合親和性を保護および回復しながら、有益な免疫回避を付与できます。
Consistent with the latest functional studies, our results showed that Q493E and F456L can act cooperatively through epistatic couplings to reverse the detrimental effect of individual Q493E mutation seen in other genetic backgrounds.
最新の機能研究と一致して、私たちの結果は、Q493E と F456L がエピスタシス結合を介して協力的に作用し、他の遺伝的背景に見られる個々の Q493E 変異の有害な影響を逆転できることを示しました。
Although precise molecular mechanisms underlying the observed epistatic effects of Q493E and F456L are likely to be complex, our results suggested that epistatic interactions between these sites may arise due to the increased side-chain flexibility of the interacting F456L, Q493E on RBD with H34 an K31 on ACE2 within a rather confined RBD-ACE2 interface.
Q493E と F456L の観察されたエピスタシス効果の根底にある正確な分子メカニズムは複雑である可能性が高いが、私たちの結果は、これらの部位間のエピスタシス相互作用は、RBD 上の Q493E と ACE2 上の H34 および K31 の相互作用する F456L の側鎖柔軟性の増加により、かなり限定された RBD-ACE2 インターフェース内で発生する可能性があることを示唆している。
The progressively increased flexibility of the RBD-ACE2 interface in the KP.2 and KP.3 complexes is manifested in multiple side-chain conformations for H34 and K31 seen in the structural ensembles as well as the enhanced plasticity of the binding interface afforded by L455S and F456L mutations.
KP.2 および KP.3 複合体における RBD-ACE2 インターフェースの柔軟性が徐々に増加していることは、構造アンサンブルで見られる H34 および K31 の複数の側鎖コンフォメーション、および L455S および F456L 変異によってもたらされる結合インターフェースの強化された可塑性に表れている。
Overall, this analysis pointed to a central role of F456L as a potential regulator of epistatic couplings but also suggests a potential role of structurally proximal Y453 site supporting the improved binding interfacial interactions in the KP.3 variant.
全体として、この分析は、F456Lがエピスタシス結合の潜在的な調節因子として中心的な役割を果たしていることを示唆しているが、KP.3変異体における結合界面相互作用の改善をサポートする構造的に近接したY453部位の潜在的な役割も示唆している。
Our results are consistent with the related study showing that Omicron variants can induce distinct dynamics and exploit epistatic interactions between sites R346, F486P, Q498, Q493 to modulate the RBD-ACE2 interface with increasing flexibility in antibody binding residues.
私たちの結果は、オミクロン変異体が明確なダイナミクスを誘導し、R346、F486P、Q498、Q493部位間のエピスタシス相互作用を利用して、抗体結合残基の柔軟性を高めながらRBD-ACE2界面を調節できることを示した関連研究と一致している。
The results also demonstrated that L455, F456 and Q493 are pronounced escape hotspots of resistance to class I antibodies.
結果はまた、L455、F456、Q493がクラスI抗体に対する耐性の顕著なエスケープホットスポットであることを示した。
We suggested that epistatic couplings between these sites may not only help to restore ACE2 binding affinity but also represent a mechanism for amplifying immune response provided by individual mutations L455S, F456L and Q493E.
これらの部位間のエピスタシス結合は、ACE2結合親和性の回復に役立つだけでなく、個々の変異L455S、F456L、Q493Eによって提供される免疫応答を増幅するメカニズムを表す可能性もあると示唆した。
These arguments are also consistent with evolutionary studies revealing strong epistasis between pre-existing substitutions in Omicron variants and antibody resistance mutations acquired during selection experiments, suggesting that epistasis can also lower the genetic barrier for antibody escape.
これらの議論は、オミクロン変異体の既存の置換と選択実験中に獲得された抗体耐性変異との間に強いエピスタシスがあることを明らかにした進化研究とも一致しており、エピスタシスによって抗体回避の遺伝的障壁も低下する可能性があることを示唆している。
Based on the correspondence between the computational results and biochemical experiments we suggest that the primary role of BA.2.86, JN.1, KP.2, and KP.3 mutations may be to ensure a broad resistance against different classes of RBD antibodies, while several important mutations such as Q493E could confer the improved ACE2 binding affinity.
計算結果と生化学実験の対応に基づいて、BA.2.86、JN.1、KP.2、および KP.3 変異の主な役割は、さまざまなクラスの RBD 抗体に対する広範な耐性を確保することである可能性があり、Q493E などのいくつかの重要な変異は、ACE2 結合親和性の向上をもたらす可能性があると示唆しています。
The results of our study provide an additional evidence to the mechanism proposed by Bloom and colleagues that recently evolving variants tend to exploit a fairly focused group of antibody-escape sites ( L455, F456, R403, D405) through convergent evolution as these positions are under moderate functional constraint and mutations incur little cost to virus.
私たちの研究結果は、最近進化した変異体が、収斂進化を通じてかなり集中した抗体回避部位のグループ(L455、F456、R403、D405)を利用する傾向があるという、ブルームと同僚によって提案されたメカニズムに対する追加の証拠を提供します。これらの位置は中程度の機能的制約を受けており、突然変異はウイルスにほとんどコストをかけません。
Although the effect of immune evasion could be more variant-dependent and modulated through recruitment of mutational sites in various adaptable RBD regions, the currently dominating variants operate on a limited number of convergent escape hotspots.
免疫回避の効果は、より変異体に依存し、さまざまな適応可能なRBD領域での突然変異部位の採用によって調整される可能性がありますが、現在支配的な変異体は、限られた数の収斂回避ホットスポットで機能します。
At the same time, the epistatic interactions between convergent mutational sites and ACE2 binding affinity hotspots may represent a viable mechanism to retain ACE2 binding with minimum resources and leveraging the immune escape centers to protect ACE2 affinity.
同時に、収斂突然変異部位とACE2結合親和性ホットスポット間のエピスタシス相互作用は、最小限のリソースでACE2結合を維持し、免疫回避センターを活用してACE2親和性を保護する実行可能なメカニズムを表している可能性があります。
5. Conclusions
In this study, we combined AF2-based atomistic predictions of structures and conformational ensembles of the SARS-CoV-2 Spike complexes with the host receptor ACE2 for the most recent dominant Omicron variants JN.1, KP.1, KP.2 and KP.3 to examine the mechanisms underlying the role of convergent evolution hotspots in balancing ACE2 binding and antibody evasion.
本研究では、最新の優勢なオミクロン変異体 JN.1、KP.1、KP.2、KP.3 について、SARS-CoV-2 スパイク複合体の構造とコンフォメーション アンサンブルの AF2 ベースの原子予測と宿主受容体 ACE2 を組み合わせて、ACE2 結合と抗体回避のバランスをとる収斂進化ホットスポットの役割の根底にあるメカニズムを調べました。
The AF2-predicted conformational ensembles suggested the increased heterogeneity in the JN.1, KP.2 and especially KP.3 RBD variants which may potentially enable these variants to leverage a more mobile RBD structure to modulate and evade antibody neutralization.
AF2 予測のコンフォメーション アンサンブルは、JN.1、KP.2、特に KP.3 RBD 変異体の不均一性の増加を示唆しており、これらの変異体がより可動性の高い RBD 構造を利用して抗体中和を調整および回避できる可能性があります。
The results showed that the majority of mutational sites remain in similar positions in different conformations of the AF2 ensembles, but convergent mutational sites R346T, L455S, F456L and Q493E as well as in their interacting ACE2 sites K31, H34, E35 may exhibit some moderate level of plasticity and display concerted conformational rearrangements at the binding interface.
結果は、AF2 アンサンブルの異なるコンフォメーションで変異部位の大部分が同様の位置に留まっているが、収束変異部位 R346T、L455S、F456L、Q493E およびそれらの相互作用する ACE2 部位 K31、H34、E35 は、ある程度の可塑性を示し、結合界面で協調的なコンフォメーション再配置を示す可能性があることを示した。
Using conformational ensembles obtained from MD simulation studies of the RBD-ACE2 complexes for the BA.2.86, JN.1, KP.2 and KP.3 variants we performed a systematic mutational scanning of the RBD residues in the RBD-ACE2 complexes.
BA.2.86、JN.1、KP.2、KP.3 変異体の RBD-ACE2 複合体の MD シミュレーション研究から得られたコンフォメーション アンサンブルを使用して、RBD-ACE2 複合体の RBD 残基の系統的な変異スキャンを実行しました。
Our results suggested that F456L and Q493E may be only marginally deleterious and may reverse their effect on ACE2 binding in a different genetic background.
結果は、F456L と Q493E はわずかに有害である可能性があり、異なる遺伝的背景では ACE2 結合に対するそれらの影響を逆転させる可能性があることを示唆しました。
Mutational profiling in the KP.3 background showed that Q493E mutation becomes more favorable when combined together with KP.3 changes L455S and especially F456L.
KP.3 背景での変異プロファイリングにより、Q493E 変異は、KP.3 の L455S および特に F456L 変異と組み合わせると、より有利になることが示されました。
The results of our study supported a recently proposed hypothesis that KP.2 and KP.3 lineage may have evolved to outcompete other Omicron subvariants by improving immune suppression while balancing binding affinity with ACE2 via through compensatory epistatic effect of L455S, F456L, Q493E and F486P mutations.
私たちの研究結果は、KP.2 および KP.3 系統が、L455S、F456L、Q493E、および F486P 変異の補償的エピスタシス効果を介して ACE2 との結合親和性のバランスを取りながら免疫抑制を改善することで、他の Omicron サブバリアントに打ち勝つように進化した可能性があるという最近提案された仮説を裏付けました。
Based on structural analysis of the conformational ensembles and MM-GBSA computations of the RBD-ACE2 complexes for JN.1, KP.2 and KP.3 variants, we argue that the plasticity of the ACE2 interfacial sites that are afforded by L455S/F456L mutations can be properly exploited by Q493E, Y453 and F456L positions to enhance binding affinity.
JN.1、KP.2、KP.3 変異体の RBD-ACE2 複合体の立体配座アンサンブルの構造解析と MM-GBSA 計算に基づいて、L455S/F456L 変異によってもたらされる ACE2 界面部位の可塑性は、Q493E、Y453、F456L の位置によって適切に利用され、結合親和性を高めることができると主張します。
Overall, this analysis pointed to a central role of F456L as a potential regulator of epistatic couplings but also suggests a potential role of structurally proximal L452 and Y453 sites supporting the improved binding interfacial interactions in the KP.3 variant.
全体として、この分析は、F456L がエピスタシス結合の潜在的な調節因子として中心的な役割を果たしていることを指摘しましたが、KP.3 変異体における結合界面相互作用の改善をサポートする構造的に近接した L452 および Y453 部位の潜在的な役割も示唆しています。
Structure-based mutational scanning of the RBD binding interfaces with different classes of RBD antibodies characterized the role of specific mutations in eliciting broad resistance to neutralization against distinct epitope classes.
さまざまなクラスの RBD 抗体による RBD 結合界面の構造ベースの変異スキャンにより、異なるエピトープ クラスに対する中和に対する広範な抵抗を誘発する特定の変異の役割が明らかになりました。
The results demonstrated that JN.1, KP.2 and KP.3 variants harboring the L455SS, F456L and Q493E mutations can significantly impair the neutralizing activity of RBD class-1 monoclonal antibodies, also revealing that Y453, L455 and F456 emerged as major escape hotspots for these variants.
結果は、L455SS、F456L、Q493E 変異を持つ JN.1、KP.2、KP.3 変異体が RBD クラス 1 モノクローナル抗体の中和活性を著しく損なう可能性があることを示し、また、Y453、L455、F456 がこれらの変異体の主要な回避ホットスポットとして出現したことも明らかにしました。
These results are consistent with the experimental data showing that antibody evasion drives the convergent evolution of L455F/S and F456L, while the epistatic interactions mediated by F456L can facilitate the favorable contribution of Q493E to restore ACE2 binding.
これらの結果は、抗体回避が L455F/S と F456L の収束進化を促進し、F456L によって媒介されるエピスタシス相互作用が Q493E の好ましい寄与を促進して ACE2 結合を回復できることを示した実験データと一致しています。
The results support the notion that evolution of Omicron variants may favor emergence of lineages with beneficial combinations of mutations involving mediators of epistatic couplings that control balance of high ACE2 affinity and immune evasion.
この結果は、オミクロン変異体の進化が、ACE2 の高い親和性と免疫回避のバランスを制御するエピスタシス結合のメディエーターを含む変異の有益な組み合わせを持つ系統の出現に有利に働く可能性があるという考えを裏付けています。
Our study provided support to a mechanism in which convergent Omicron mutations can promote high transmissibility and antigenicity of the virus by controlling the interplay between the binding to the host receptor and robust immune evasion profile.
私たちの研究は、収束的なオミクロン変異が、宿主受容体への結合と強力な免疫回避プロファイルの相互作用を制御することで、ウイルスの高い伝染性と抗原性を促進できるメカニズムを裏付けました。
This may potentially be a continuous common strategy of Omicron evolution that would result in combinatorial exploration of convergent mutations to evade neutralizing antibodies and while still maintaining ACE2 binding affinity.
これは、中和抗体を回避しながらも ACE2 結合親和性を維持する収束的な変異の組み合わせの探索につながる、オミクロン進化の継続的な共通戦略である可能性があります。
以下省略。
この記事が気に入ったらサポートをしてみませんか?