SARS-CoV-2スパイクタンパク質の構造生物学とそれが免疫システムを欺く方法

Up, up, down, down: the structural biology of the SARS-CoV-2 spike protein and how it cheats the immune system
上、上、下、下:SARS-CoV-2スパイクタンパク質の構造生物学とそれが免疫システムを欺く方法
Abstract
Spike proteins protrude from the SARS-CoV-2 viral envelope and are responsible for initiating fusion into human epithelial cells by binding Angiotensin-Converting Enzyme 2 receptors on the host cell surface.
スパイクタンパク質は、SARS-CoV-2ウイルスエンベロープから突出しており、宿主細胞表面のアンジオテンシン変換酵素2受容体に結合することでヒト上皮細胞への融合を開始する役割を果たします。
Due to their exposed location on the outside of the virion and their key role in infection, SARS-CoV-2 spike proteins are an important target for vaccine development and drug design.
ウイルス粒子の外側に露出しており、感染において重要な役割を果たすため、SARS-CoV-2スパイクタンパク質はワクチン開発と薬剤設計の重要なターゲットです。
Over the last two years, many spike protein structures have been experimentally determined, providing essential details into the complex structural rearrangements that occur after receptor binding and during fusion of the virion with the host cell, as well as into the interactions of spike protein molecules with antibodies.
過去 2 年間で、多くのスパイクタンパク質構造が実験的に決定され、受容体結合後およびウイルス粒子と宿主細胞の融合中に発生する複雑な構造再配置、およびスパイクタンパク質分子と抗体の相互作用に関する重要な詳細が得られました。
SARS-CoV-2 variants, particularly those associated with reduced vaccine efficacy, are strongly associated with mutations in two domains of the SARS-CoV-2 spike protein, namely the receptor binding domain and the N-terminal domain, which have both been structurally characterized.
SARS-CoV-2 変異体、特にワクチンの有効性の低下に関連する変異体は、SARS-CoV-2 スパイクタンパク質の 2 つのドメイン、つまり受容体結合ドメインと N 末端ドメインの変異と強く関連しており、どちらも構造的に特徴付けられています。
This review provides a comprehensive overview of the structural knowledge acquired over the past four years on the SARS-CoV-2 spike protein and its critical role in viral infection.
このレビューでは、過去 4 年間に得られた SARS-CoV-2 スパイクタンパク質の構造に関する知識と、ウイルス感染におけるその重要な役割について包括的な概要を示します。
1. Introduction
The infection caused by the novel severe acute respiratory syndrome coronavirus SARS-CoV-2 was declared a pandemic by the WHO in March 2020, and continues to be a serious health concern worldwide to this day.
新型重症急性呼吸器症候群コロナウイルスSARS-CoV-2による感染症は、2020年3月にWHOによってパンデミックと宣言され、現在も世界中で深刻な健康問題となっています。
Coronaviruses are named for the crown-like appearance of glycosylated fusion proteins (called ‘spike’ proteins, S proteins or surface glycoproteins) protruding from the surface of each virion.
コロナウイルスは、各ウイルス粒子の表面から突き出ている糖化融合タンパク質(「スパイク」タンパク質、Sタンパク質、または表面糖タンパク質と呼ばれる)の王冠のような外観にちなんで名付けられています。
In this review, the word ‘spike’ will always refer to the entire complex from SARS-CoV-2 including its glycosylation, while the protein itself is called ‘spike protein’ and one chain of spike protein in the context of the spike will be identified at a ‘spike protomer’.
このレビューでは、「スパイク」という言葉は常に、グリコシル化を含む SARS-CoV-2 の複合体全体を指しますが、タンパク質自体は「スパイクタンパク質」と呼ばれ、スパイクの文脈におけるスパイクタンパク質の 1 つの鎖は「スパイクプロトマー」で識別されます。
Spikes are trimeric and extend approximately 20 nm from the viral surface membrane.
スパイクは三量体であり、ウイルス表面膜から約 20 nm 伸びています。
Observed virions contain small numbers of spikes (one study reports 26 ± 15), and far fewer than previously observed for SARS-CoV-1.
観察されたウイルス粒子には少数のスパイク(1つの研究では26±15と報告)が含まれており、SARS-CoV-1で以前に観察された数よりもはるかに少ない。
Spikes initiate entry into human host cells by interacting with the cell surface receptor Angiotensin-Converting Enzyme 2 (ACE2).
スパイクは、細胞表面受容体であるアンジオテンシン変換酵素 2 (ACE2) と相互作用することで、ヒト宿主細胞への侵入を開始します。
ACE2 is mainly expressed in lung and intestinal epithelial cells, but is also present on kidney, cardiovascular and gastrointestinal cells.
ACE2 は主に肺と腸の上皮細胞で発現していますが、腎臓、心血管、胃腸の細胞にも存在します。
After engaging ACE2, the spike is likely processed by proteases and parts of the complex dissociate before the remaining parts undergo major structural rearrangements necessary for membrane fusion.
ACE2 と結合した後、スパイクはおそらくプロテアーゼによって処理され、複合体の一部が解離してから、残りの部分が膜融合に必要な主要な構造再配置を受けると考えられます。
As part of the refolding mechanism, the spike undergoes an irreversible transformation into a trimeric hairpin structure.
リフォールディング機構の一環として、スパイクは不可逆的な三量体ヘアピン構造への変形を受ける。
This structural change brings the viral and cellular cell membranes into close proximity, ultimately initiating membrane fusion.
この構造変化により、ウイルスと細胞膜が接近し、最終的に膜融合が始まります。
Important to note is that the precise mechanism by which a SARS-CoV-2 virion fuses with the host cell is not yet fully understood and our understanding relies mostly on research of other fusion proteins.
注目すべき重要な点は、SARS-CoV-2 ウイルス粒子が宿主細胞と融合する正確なメカニズムはまだ完全には解明されておらず、私たちの理解は主に他の融合タンパク質の研究に依存しているということです。
Once the membranes are fused, the viral RNA is released into the cytosol of the host cell and its first open reading frame is translated by the host ribosome machinery to create the two variants of the viral polyprotein.
膜が融合すると、ウイルスRNAが宿主細胞の細胞質に放出され、その最初のオープンリーディングフレームが宿主リボソーム機構によって翻訳され、ウイルスポリタンパク質の2つの変異体が生成されます。
Thus begins the cascade of events that results in the production of new viral particles.
こうして、新しいウイルス粒子の生成につながる一連のイベントが始まります。
The spike is thereby key to invading the host cell and therefore crucial to the viral infection cycle.
スパイクは宿主細胞への侵入の鍵となるため、ウイルス感染サイクルにとって極めて重要です。
Due to their exposed position on the viral surface membrane, spikes are also a major target for the host’s immune system and an important target for drug and vaccine development.
スパイクはウイルス表面膜に露出しているため、宿主の免疫システムの主要な標的であり、薬剤やワクチン開発の重要な標的でもあります。
However, as a way of reducing recognition by antibodies of the immune system, the surfaces of spikes are densely covered with complex carbohydrates, primarily N-glycans.
しかし、免疫系の抗体による認識を減らす方法として、スパイクの表面は複合炭水化物、主に N-グリカンで密に覆われています。
The glycan shield, which masks between 60% to 90% of each spike's surface from antibodies, plays a crucial role in immune system evasion for exposed viral proteins.
各スパイクの表面の 60% ~ 90% を抗体から保護するグリカン シールドは、露出したウイルス タンパク質の免疫システム回避に重要な役割を果たします。
The attached glycans also aid the stability and maintain optimal activity of spikes.
付着したグリカンはスパイクの安定性を助け、最適な活性を維持します。
2. Structural overview
The spike protein is one of four structural proteins encoded on the SARS-CoV-2 genome, with the others being a membrane glycoprotein (M), a nucleocapsid protein (N) and the small envelope protein (E), all four of which are present in the virion.
スパイクタンパク質は、SARS-CoV-2ゲノムにコード化された4つの構造タンパク質の1つで、他の2つは膜糖タンパク質(M)、ヌクレオカプシドタンパク質(N)、小型エンベロープタンパク質(E)であり、これら4つはすべてウイルス粒子に存在します。
The spike protein is encoded in the gene region between 21,563 and 25,384 bp, and is the largest of the four structural proteins ranging from 1250 to 1273 amino acids in length (Figure 1a), with the exact sequence and length depending on the virus strain.
スパイクタンパク質は、21,563~25,384bpの遺伝子領域にコード化されており、長さが1250~1273アミノ酸の範囲にある4つの構造タンパク質の中で最大です(図1a)。正確な配列と長さはウイルス株によって異なります。
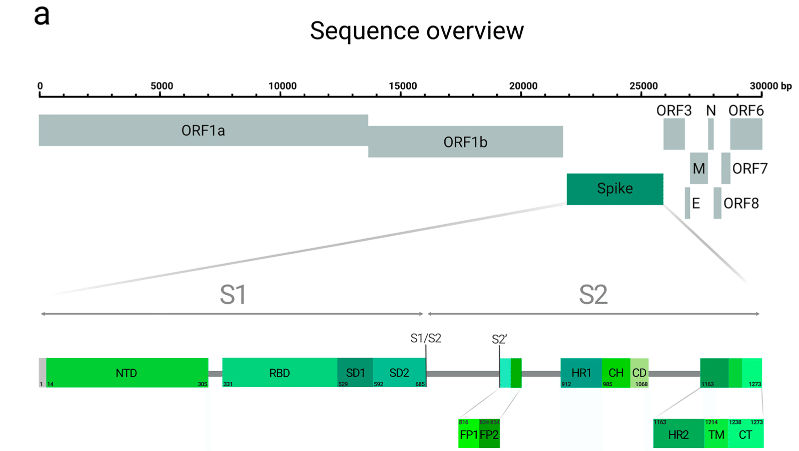
As a class I fusion protein, spike proteins form homotrimers, with each protomer comprising a large extracellular domain (EC) at the N-terminal end, a single α-helical transmembrane anchor (TM), and a short intracellular cytoplasmic tail (CT) at the C-terminus.
クラス I 融合タンパク質であるスパイクタンパク質はホモ三量体を形成し、各プロトマーは N 末端の大きな細胞外ドメイン (EC)、単一の α ヘリカル膜貫通アンカー (TM)、および C 末端の短い細胞内細胞質テール (CT) で構成されます。
The extracellular part of the spike protein can be divided into two subunits, S1 and S2 (Table 1): Subunit S1 (also called the ‘head’) is responsible for receptor binding while S2 (the ‘stalk’) mediates membrane fusion.
スパイクタンパク質の細胞外部分は、S1 と S2 の 2 つのサブユニットに分けられます (表 1)。サブユニット S1 (「ヘッド」とも呼ばれる) は受容体結合を担い、S2 (「ストーク」) は膜融合を媒介します。
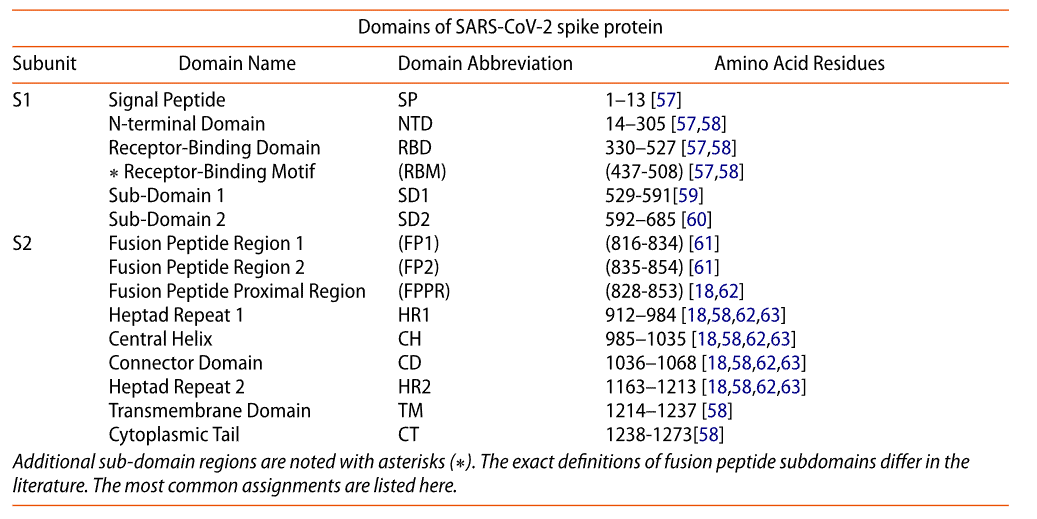
S1, the first 685 amino acids, is further subdivided into five segments: a short signal peptide (SS), the N-terminal domain (NTD), the receptor-binding domain (RBD), and two subdomains (SD1 and SD2).
最初の 685 個のアミノ酸である S1 は、さらに 5 つのセグメントに分割されます。短いシグナル ペプチド (SS)、N 末端ドメイン (NTD)、受容体結合ドメイン (RBD)、および 2 つのサブドメイン (SD1 と SD2) です。
The S2 subunit, encompassing 588 amino acid residues, is subdivided into an N-terminal hydrophobic fusion peptide (FP), two heptad repeats (HR1 and HR2), the transmembrane domain and cytoplasmic tail (Figure 1a).
588 個のアミノ酸残基を含む S2 サブユニットは、N 末端疎水性融合ペプチド (FP)、2 つのヘプタド リピート (HR1 と HR2)、膜貫通ドメイン、および細胞質テールに分割されます (図 1a)。
2.1. Subunit S1
The S1 subunit, which binds Angiotensin-Converting Enzyme 2, comprises a signal peptide (SP) and four domains, listed in Table 1.
アンジオテンシン変換酵素 2 に結合する S1 サブユニットは、シグナル ペプチド (SP) と 4 つのドメインで構成されており、表 1 にリストされています。
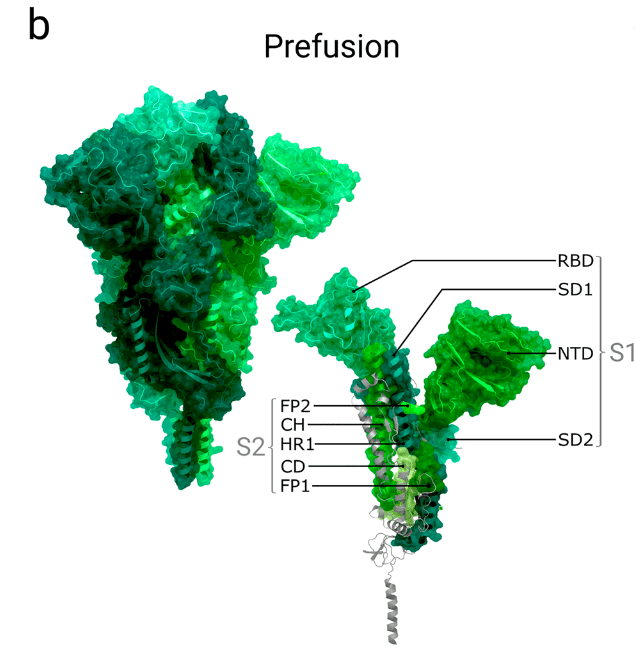
These four domains (of each S1 monomer) wrap around and cap the top of the helical S2 subunit (Figure 1b).
これらの 4 つのドメイン (各 S1 モノマー) は、らせん状の S2 サブユニットの上部を包み込み、キャップします (図 1b)。
The spike protein is synthesized on the rough endoplasmic reticulum before being transported to the Golgi complex ; the targeting to the rough endoplasmic reticulum is done by the N-terminal signal peptide (SP), which consists of thirteen amino acids with helix-forming, highly hydrophobic residues.
スパイクタンパク質は、ゴルジ体へ輸送される前に粗面小胞体で合成されます。粗面小胞体への標的化は、らせん状で疎水性の高い残基を持つ13個のアミノ酸からなるN末端シグナルペプチド(SP)によって行われます。
Co-translational and post-translational processing result in the removal of this signal peptide in the ER lumen, trimerization of the spike protein, extensive glycosylation, and subunit cleavage by furin or furin-like proteases.
翻訳時および翻訳後の処理により、ER 内腔でのこのシグナルペプチドの除去、スパイクタンパク質の三量体化、広範なグリコシル化、およびフーリンまたはフーリン様プロテアーゼによるサブユニットの切断が起こります。
The N-terminal domain is similar to MERS- and bovine-CoV functionalized with eight N-linked glycan molecules.
N末端ドメインは、8つのN結合グリカン分子で機能化されたMERS-CoVおよびウシCoVに類似しています。
It shares only 52% sequence identity with SARS-CoV-1, and is highly similar to the equivalent domain in bat coronavirus RatG13 (>98%).
SARS-CoV-1との配列同一性はわずか52%で、コウモリコロナウイルスRatG13の同等のドメインと非常に類似しています(>98%)。
The N-terminal domain can be divided into three topological regions: top, core and bottom.
N末端ドメインは、トップ、コア、ボトムの3つのトポロジカル領域に分けることができます。
The exact role of the N-terminal domain is still not fully understood, but has been suggested that the sequence 72-GTNGTKR-78 (located in a loop connecting two core-region β-sheets), carrying a highly conserved N-glycosylation site at N74, can bind host-cell saccharides and/or additional receptors aside from ACE2.
N末端ドメインの正確な役割はまだ完全には解明されていないが、N74に高度に保存されたNグリコシル化部位を持つ72-GTNGTKR-78配列(2つのコア領域βシートを接続するループに位置する)は、ACE2以外の宿主細胞糖類および/または追加の受容体に結合することができると示唆されている。
Receptor binding through the N-terminal domain (NTD) has been previously observed in spike proteins of the mouse hepatitis virus, a member of the Betacoronaviridae family.
N末端ドメイン(NTD)を介した受容体結合は、ベータコロナウイルス科に属するマウス肝炎ウイルスのスパイクタンパク質で以前に観察されています。
Additionally, the spike protein’s N-terminal domain may also be involved in interaction with the host cells via recognition of specific sugar molecules, as occurs in the transmissible gastroenteritis coronavirus and bovine coronavirus.
さらに、スパイクタンパク質のN末端ドメインは、伝染性胃腸炎コロナウイルスや牛コロナウイルスで見られるように、特定の糖分子の認識を介して宿主細胞との相互作用にも関与している可能性がある。
Structures of the prefusion spikes are often characterised by the conformation of their receptor binding domain (RBDs).
融合前スパイクの構造は、多くの場合、受容体結合ドメイン (RBD) の立体構造によって特徴付けられます。
In the trimeric spike, each RBD is located next to the N-terminal domain of the adjacent protomer and can adopt an ‘up’ and a ‘down’ conformation.
三量体スパイクでは、各 RBD は隣接するプロトマーの N 末端ドメインの隣に位置し、「上向き」および「下向き」の立体構造をとることができます。
It is in the ‘up’ state that the receptor binding domain interacts with host cell surface ACE2 receptors.
受容体結合ドメインが宿主細胞表面の ACE2 受容体と相互作用するのは、「上向き」の状態です。
The RBD consists of a five-stranded antiparallel β-sheet core with short connecting α-helices, β-sheets and loops.
RBD は、5 本の鎖からなる平行な β シート コアと、短い α ヘリックス、β シート、ループで構成されています。
The receptor-binding motif (RBM) is located as an extended insertion between two β-strands and contains most of the residues which mediate ACE2 binding.
受容体結合モチーフ (RBM) は、2 つの β 鎖の間に挿入された延長部分として位置し、ACE2 結合を媒介する残基のほとんどを含んでいます。
The RBD is stabilized by four disulphide bonds, three of which are located in the core (Cys336–Cys361, Cys379–Cys432, Cys391–Cys525), with the last stabilizing loops at the distal end of the RBM (Cys480-Cys488).
RBD は 4 つのジスルフィド結合によって安定化されており、そのうち 3 つはコア (Cys336–Cys361、Cys379–Cys432、Cys391–Cys525) にあり、最後の安定化ループは RBM の遠位端 (Cys480-Cys488) にあります。
Both subdomains SD1 and SD2 (sometimes called C-terminal domain 1 and 2 or CTD1 and CTD2) are β-structure rich segments located after the RBD.
サブドメイン SD1 と SD2 (C 末端ドメイン 1 と 2、または CTD1 と CTD2 と呼ばれることもある) は両方とも、RBD の後に位置する β 構造に富んだセグメントです。
The N-terminal end of S2 is adjacent to a cleavage site targeted by furin (or another protease) during pre-processing of the spike protein.
S2 の N 末端は、スパイクタンパク質の前処理中にフーリン (または別のプロテアーゼ) によって標的とされる切断部位に隣接しています。
2.2. Subunit S2
The central ‘stalk’ of the spike protein, responsible for viral fusion and entry, is formed of seven domains, listed in Table 1.
スパイクタンパク質の中央の「茎」は、ウイルスの融合と侵入を担い、表 1 に示す 7 つのドメインで構成されています。
The helical core of the S2 subunit is formed by the three-stranded coiled coil central helix domain, with most of the remaining polypeptide chain, including heptad repeat 1, the connector domain and the fusion peptide, wrapping around it (Figure 1b).
S2 サブユニットのらせん状のコアは、3 本鎖のコイルドコイル中央ヘリックスドメインによって形成され、残りのポリペプチド鎖の大部分 (ヘプタド繰り返し 1、コネクタドメイン、融合ペプチドを含む) がその周囲を包み込んでいます (図 1b)。
HR1 and HR2 are characterized by seven-residue repeats of the sequence HPPHCPC in tandem (where H stands for a bulky hydrophobic residue, P is a polar or hydrophilic residue and C is any charged residue), which are important during later pre-fusion rearrangements (described later).
HR1 と HR2 は、HPPHCPC 配列の 7 残基の繰り返しが直列に並んでいることを特徴としています (H はかさばる疎水性残基、P は極性または親水性残基、C は任意の荷電残基)。これらは、後続の融合前再配置 (後述) で重要になります。
The exact sequence of the fusion peptide regions (FP1 and 2), which are inserted into the host cell membrane during the fusion process, have not yet been definitively determined.
融合プロセス中に宿主細胞膜に挿入される融合ペプチド領域 (FP1 および 2) の正確な配列はまだ決定されていません。
The proposed fusion peptide regions encompassing FP1 and FP2 contain approximately 15–20 predominantly hydrophobic amino acids, and are located downstream to the S2’ cleavage site at R815-S816.
FP1 と FP2 を含む提案された融合ペプチド領域には、主に疎水性のアミノ酸が約 15~20 個含まれており、S2’ 切断部位の下流の R815-S816 に位置しています。
FP1 and FP2 were determined based on their relatively well-conserved sequences.
FP1 と FP2 は、比較的よく保存された配列に基づいて決定されました。
FP1 comprises a conserved hydrophobic LLF motif (Leu821-Leu822-Phe823) while FP2 features a pair of highly conserved cysteine residues which both seem to play an important role during membrane fusion.
FP1 は保存された疎水性 LLF モチーフ (Leu821-Leu822-Phe823) で構成され、FP2 は高度に保存されたシステイン残基のペアを特徴としており、どちらも膜融合時に重要な役割を果たすようです。
The transmembrane domain (TM) is highly conserved in all coronaviruses.
膜貫通ドメイン(TM)は、すべてのコロナウイルスで高度に保存されています。
It contains three regions: an aromatic region, a hydrophobic central region, and a cysteine-rich region.
これには、芳香族領域、疎水性中央領域、およびシステインに富む領域の 3 つの領域が含まれます。
The central hydrophobic part of the transmembrane domain forms an α-helix that arranges as a trimer with its counterparts.
膜貫通ドメインの中央の疎水性部分は、対応する部分と三量体として配置されるαヘリックスを形成します。
Together with the spike protein’s cytoplasmic tail (CT), the transmembrane domain helps to stabilize the trimeric structure that is crucial for membrane fusion.
スパイクタンパク質の細胞質テール(CT)とともに、膜貫通ドメインは、膜融合に不可欠な三量体構造を安定化するのに役立ちます。
A study of the related coronavirus SARS-CoV-1 revealed that the rate of infection can be decreased by replacing hydrophobic residues in the central hydrophobic transmembrane section by hydrophilic ones such as lysine.
関連するコロナウイルス SARS-CoV-1 の研究では、中央の疎水性膜貫通セクションの疎水性残基をリジンなどの親水性残基に置き換えることで感染率を低下させることができることが明らかになりました。
Replacing cysteine residues in the TM domain proximal to the membrane, next to the central hydrophobic part, inhibits membrane fusion completely whereas mutation of other cysteines in the cysteine-rich region does not have an impact on fusion.
膜に近い TM ドメインのシステイン残基を中央の疎水性部分に隣接する部分に置き換えると、膜融合が完全に阻害されますが、システインに富む領域内の他のシステインを変異させても融合には影響しません。
In all currently available experimentally determined prefusion structures, the heptad repeat 2 domain, transmembrane domain and the cytoplasmic tail are not resolved.
現在入手可能な実験的に決定された融合前構造のすべてにおいて、ヘプタッドリピート2ドメイン、膜貫通ドメイン、および細胞質テールは解明されていない。
The cytoplasmic tail (CT) of coronavirus spike proteins is known to contain localization signals for the endoplasmic reticulum-Golgi intermediate compartment.
コロナウイルススパイクタンパク質の細胞質テール(CT)には、小胞体ゴルジ体中間区画の局在シグナルが含まれていることが知られている。
At the farthest C-terminal end, a novel dibasic motif within the sequence KLHYT (KXHXX) has been identified as a functional ER retrieval signal, where X denotes any amino acid residue.
最も遠い C 末端では、配列 KLHYT (KXHXX) 内の新しい二塩基モチーフが機能的な ER 回収シグナルとして特定されています。ここで、X は任意のアミノ酸残基を示します。
This motif may also contribute to interactions of the spike protein with copies of the membrane (M) protein located in the virion envelope In both SARS-CoV-1 and SARS-CoV-2, the folded cytoplasmic tail domain may interact with the M protein to facilitate incorporation of a complete spike into the new virus particle.
このモチーフは、スパイクタンパク質と、ウイルス粒子エンベロープにある膜(M)タンパク質のコピーとの相互作用にも寄与している可能性があります。SARS-CoV-1とSARS-CoV-2の両方で、折り畳まれた細胞質テールドメインがMタンパク質と相互作用して、新しいウイルス粒子への完全なスパイクの組み込みを促進する可能性があります。
2.3. Availability and consistency of structures of the spike
Previous outbreaks of related viruses, namely of SARS-CoV-1 in China and Canada in February 2003, and of the Middle East respiratory syndrome coronavirus (MERS-CoV) since April 2012, lead to depositions of 43 SARS-CoV–1 and 29 MERS-CoV structures of the spike in the Protein Data Bank (PDB).
関連するウイルスの過去の流行、すなわち2003年2月に中国とカナダで発生したSARS-CoV-1、および2012年4月以降の中東呼吸器症候群コロナウイルス(MERS-CoV)の流行により、タンパク質データバンク(PDB)に43のSARS-CoV-1および29のMERS-CoVスパイク構造が登録されました。
Their availability accelerated scientific progress at the beginning of the COVID-19 pandemic in 2020, as the spikes of SARS-CoV-2 are highly similar to those from SARS-CoV-1 and MERS-CoV; the spike protein of SARS-CoV-2 has up to 76% sequence identity and 86% sequence similarity with that of SARS-CoV-1, depending on the viral strain.
2020年のCOVID-19パンデミックの初期には、SARS-CoV-2のスパイクがSARS-CoV-1やMERS-CoVのスパイクと非常に類似していることから、その利用可能性は科学の進歩を加速させました;SARS-CoV-2のスパイクタンパク質は、ウイルス株によって異なりますが、SARS-CoV-1のスパイクタンパク質と最大76%の配列同一性と86%の配列類似性があります。
However it has an even closer relationship with the spike protein of RaTG13, a SARS-like beta coronavirus found in bats, with up to 97.4% sequence identity and 98.4% sequence similarity, indicating a close phylogenetic relationship between the two viruses.
しかし、コウモリに見られるSARSのようなベータコロナウイルスであるRaTG13のスパイクタンパク質とはさらに密接な関係があり、最大97.4%の配列同一性と98.4%の配列類似性があり、2つのウイルスの間に密接な系統関係があることを示しています。
RaTG13 is less studied, with only three structures of its spike present in the PDB.
RaTG13はあまり研究されておらず、そのスパイクの構造はPDBに3つしかありません。
Comparison of viral spike structures from SARS-CoV-2 (PDB ID: 6VSB) and SARS-CoV-1 (PDB ID: 6CRZ) in a recent publication, gives a global Cα r.m.s.d. of 3.8 Å, indicating that both structures are broadly similar with their receptor binding domains in the ‘down’ conformation.
最近の出版物で SARS-CoV-2 (PDB ID: 6VSB) と SARS-CoV-1 (PDB ID: 6CRZ) のウイルススパイク構造を比較すると、全体的な Cα r.m.s.d. は 3.8 Å となり、両方の構造が受容体結合ドメインが「ダウン」コンフォメーションにある点で概ね類似していることを示しています。
One of the more significant differences is that in SARS-CoV-1, the receptor binding domain (RBD) packs more tightly against the N-terminal domain of the neighbouring protomer, as opposed to SARS-CoV-2 where it is located closer to the trimer’s central cavity.
より重要な違いの 1 つは、SARS-CoV-1 では受容体結合ドメイン (RBD) が隣接するプロトマーの N 末端ドメインに対してより密集しているのに対し、SARS-CoV-2 では三量体の中央空洞に近い位置にあることです。
Comparing individual domains and subunits shows a higher degree of structural homology with Cα r.m.s.d. values of 2.6 Å (NTD), 3.0 Å (RBD), 2.7 Å (SD1 and SD2) and 2.0 Å (S2 subunit).
個々のドメインとサブユニットを比較すると、Cα r.m.s.d. 値が 2.6 Å (NTD)、3.0 Å (RBD)、2.7 Å (SD1 および SD2)、2.0 Å (S2 サブユニット) と、構造相同性が高いことがわかります。
Another analysis compared only the receptor binding domains of both viruses (SARS-CoV-2: PDB ID 6M0J; SARS-CoV-1: PDB ID 2AJF), finding a lower Cα r.m.s.d. of 1.2 Å.
別の分析では、両ウイルスの受容体結合ドメインのみを比較し(SARS-CoV-2:PDB ID 6M0J、SARS-CoV-1:PDB ID 2AJF)、Cα r.m.s.d. が 1.2 Å と低いことがわかりました。
The receptor binding motifs show a higher sequence variation but still high structural conservation (1.3 Å Cα r.m.s.d.), indicating that the overall binding mode between SARS-CoV-1 and SARS-CoV-2 is nearly identical.
受容体結合モチーフは、配列の変異が大きいものの、構造の保存性は依然として高く(1.3 Å Cα r.m.s.d.)、SARS-CoV-1 と SARS-CoV-2 の全体的な結合モードはほぼ同じであることを示しています。
There are a number of other available services and studies of coronavirus structures, including specific analysis of the spike protein variability, and resources for accumulating data beyond experimental structures (https://covid.molssi.org/).
コロナウイルス構造に関する利用可能なサービスや研究は他にも多数あり、スパイクタンパク質の変動性の特定の分析や、実験構造を超えたデータを蓄積するためのリソースなどがあります。
Due to its key role in host-cell invasion, the spike is one of the most researched complexes of SARS-CoV-2 and one of the most common structures in the PDB: in total, more than 800 structures have been deposited.
スパイクは宿主細胞への侵入において重要な役割を果たすため、SARS-CoV-2 の最も研究されている複合体の 1 つであり、PDB で最も一般的な構造の 1 つです。合計で 800 を超える構造が登録されています。
Structure-solution by single-particle cryo-electron microscopy and cryo-electron tomography has provided structures of both unbound trimers (e.g. PDB 6XR8, 6X6P) and trimers in complex with receptor domains and antibodies (e.g. PDB 7EH5, 7A97, 7L2F).
単粒子クライオ電子顕微鏡法とクライオ電子トモグラフィーによる構造解析により、結合していない三量体 (例: PDB 6XR8、6X6P) と受容体ドメインおよび抗体と複合体を形成した三量体 (例: PDB 7EH5、7A97、7L2F) の両方の構造が提供されています。
Currently, there are over 600 cryo-EM structures available in the PDB ranging in resolution from 2.2 to 13.5 Å.
現在、PDB には 2.2 Å から 13.5 Å の解像度で 600 を超えるクライオ電子顕微鏡構造が登録されています。
The remaining structures have mostly been solved by X-ray crystallography, and are primarily of isolated receptor binding domains binding to Angiotensin-Converting Enzyme 2 receptor domains (e.g. PDB 6LZG), antibodies or small molecules, and range from 1.24 to 4.35 Å resolution.
残りの構造は、ほとんどがX線結晶構造解析によって解明されており、主にアンジオテンシン変換酵素2受容体ドメイン(例:PDB 6LZG)、抗体、または小分子に結合する単離受容体結合ドメインであり、解像度は1.24~4.35Åです。
NMR has been used to determine the structure of the transmembrane domain, the membrane-bound Heptad Repeat 1 domain as well as an fusion peptide domain inserted into a bilayered micelle.
NMR は、膜貫通ドメイン、膜結合型ヘプタッドリピート 1 ドメイン、および二重層ミセルに挿入された融合ペプチドドメインの構造を決定するために使用されています。
A summary of the most important experimentally determined structures available in the PDB is given in Table 2.
PDB で利用可能な最も重要な実験的に決定された構造の概要を表 2 に示します。
We highlight one story which demonstrates the incompleteness of structural models, and how users of the PDB should be careful and aware of what is not represented/resolved in the structure:
ここでは、構造モデルの不完全性、および PDB のユーザーが構造で表現/解決されていないものに注意して認識する必要があることを示す 1 つの事例を取り上げます:
The first model of a complete SARS-CoV-2 virion, published by the Centers for Disease Control and Prevention in April 2020, was created using a PDB structure that was believed to show the complete ectodomain of the spike.
2020 年 4 月に米国疾病予防管理センターによって公開された完全な SARS-CoV-2 ウイルス粒子の最初のモデルは、スパイクの完全なエクトドメインを示すと考えられていた PDB 構造を使用して作成されました。
The selected PDB structure only contained the head and the top part of the stalk of the spike molecule instead of a full-length ectodomain, and consequently, the first 3D model of a SARS-CoV-2 viral particle erroneously shows truncated spikes without full stalks; this error has been subsequently corrected but images of the incorrect model are still commonly published.
選択された PDB 構造には、完全な外部ドメインではなく、スパイク分子の頭部と柄の上部のみが含まれていたため、SARS-CoV-2 ウイルス粒子の最初の 3D モデルでは、完全な柄のない切断されたスパイクが誤って表示されています;このエラーはその後修正されましたが、誤ったモデルの画像は今でも一般的に公開されています。
To date, all experimentally determined structures of the spike show the ectodomain with only the top of stalk resolved.
現在までに、実験的に決定されたスパイク構造はすべて、茎の先端のみが解像されたエクトドメインを示しています。
Some components of the stalk, including the Heptad Repeat 2 domain, the transmembrane domain and the cytosolic tail have never been resolved in a prefusion structure, while conversely parts of the fusion peptide have only ever been resolved in prefusion state models.
ヘプタッドリピート2ドメイン、膜貫通ドメイン、細胞質テールを含む茎の一部の構成要素は、融合前構造では解像されたことがなく、逆に融合ペプチドの一部は融合前状態モデルでのみ解像されています。
Other available structures refer only to single domains or the fusion core (e.g. PDB 6M1 V and 7L4Z).
その他の利用可能な構造は、単一ドメインまたは融合コアのみを参照しています(例:PDB 6M1 Vおよび7L4Z)。
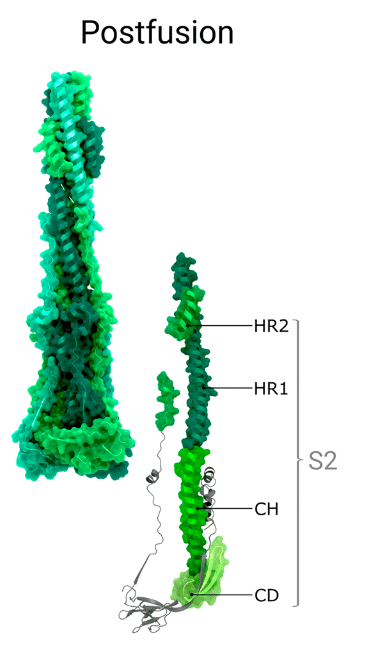
As of writing this review, there is only one experimental structure in the PDB that shows nearly the full-length post-fusion spike (Figure 1c), which was obtained via cryo–EM (PDB: 6XRA).
このレビューを書いている時点で、PDBには、ほぼ全長の融合後スパイクを示す実験構造が1つだけあります(図1c)。これは、クライオ電子顕微鏡(PDB:6XRA)で取得されました。
3. Structural changes in the spike protein during maturation and viral entry to the cell
成熟と細胞へのウイルス侵入中のスパイクタンパク質の構造変化
SARS-CoV-2 cell entry begins with the binding of spikes to angiotensin converting enzyme 2 (ACE2) receptors.
SARS-CoV-2 の細胞侵入は、スパイクがアンジオテンシン変換酵素 2 (ACE2) 受容体に結合することから始まります。
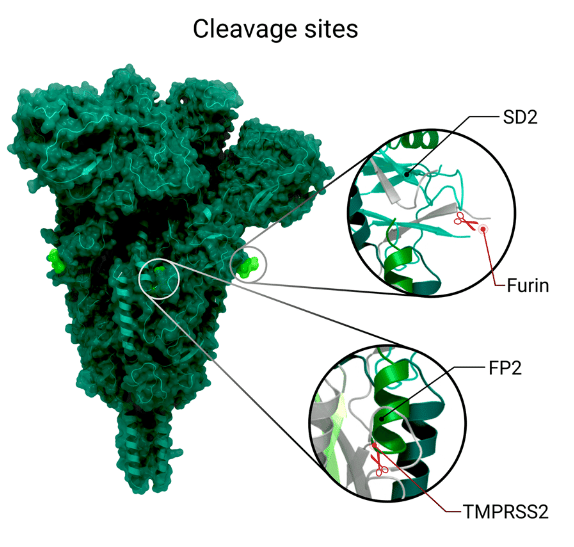
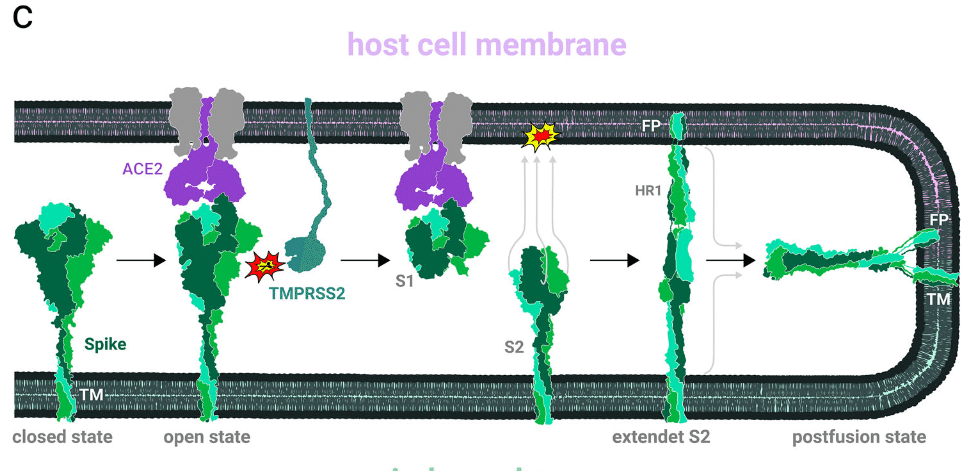
It is suggested that upon engaging the ACE2 receptor, the fusion protein undergoes proteolytic cleavage at the ‘S2’ site’ (Figure 2b), which may facilitate the release of the fusion peptides.
ACE2 受容体と結合すると、融合タンパク質は「S2」部位でタンパク質分解切断を受け(図 2b)、融合ペプチドの放出が促進される可能性があると考えられています。
The first subunit is shed, while the second subunit undergoes significant structural rearrangements to orient the fusion peptide towards and embed it into the host cell membrane.
最初のサブユニットは分離され、2 番目のサブユニットは構造的に大きく再配置され、融合ペプチドを宿主細胞膜に向け、埋め込みます。
Further rearrangements of the S2 subunit result in the formation of a hairpin structure that brings the viral and host cell membranes into close apposition, ultimately leading to membrane fusion.
S2 サブユニットのさらなる再配置によりヘアピン構造が形成され、ウイルスと宿主細胞膜が密着し、最終的に膜融合が起こります。
It is important to note that the precise mechanism of viral entry is not yet fully understood, and many steps described herein are inferred to occur based on analogy with other, well studied, viral fusion proteins such as spikes from HIV, influenza or MERS-CoV.
ウイルス侵入の正確なメカニズムはまだ完全には解明されておらず、ここで説明する多くのステップは、HIV、インフルエンザ、またはMERS-CoVのスパイクなど、よく研究されている他のウイルス融合タンパク質との類似性に基づいて発生すると推測されていることに注意することが重要です。
3.1. Spike maturation
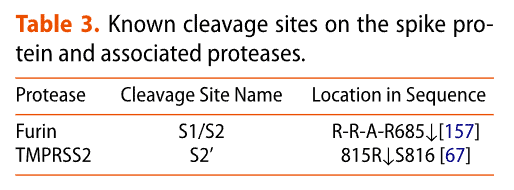
For effective infection, the spike has been suggested to mature with a proteolytic cleavage event at the S1/S2 interface during synthesis of the virus (Table 3 and Figure 2b).
効果的な感染のために、スパイクはウイルスの合成中に S1/S2 界面でタンパク質分解切断イベントを起こして成熟すると示唆されている (表 3 および図 2b)。
In contrast to the coronavirus SARS-CoV-1, the interface of the S1/S2 subunit in SARS-CoV-2 contains a multibasic cleavage site (Table 3), the existence of which is a central virulence factor for many viruses.
コロナウイルス SARS-CoV-1 とは対照的に、SARS-CoV-2 の S1/S2 サブユニットのインターフェースには多塩基切断部位 (表 3) が含まれており、その存在は多くのウイルスにとって中心的な毒性因子です。
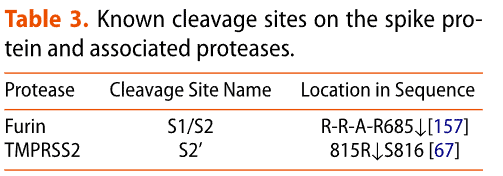
The multibasic site exhibits a RRAR amino acid motif, which resembles the canonical recognition sequence RX(R/K)R for furin, a calcium-dependent protease (X can be any amino acid residue).
多塩基部位は RRAR アミノ酸モチーフを示し、これはカルシウム依存性プロテアーゼであるフリンの標準的な認識配列 RX(R/K)R に似ています (X は任意のアミノ酸残基です)。
Cleavage in the S1/S2 site by either furin or other proteases likely happens in the Golgi apparatus of the host cell during spike synthesis.
S1/S2 部位でのフーリンまたは他のプロテアーゼによる切断は、スパイク合成中に宿主細胞のゴルジ装置で起こる可能性が高い。
After the cleavage at S1/S2, every spike protomer consists of two subunits that remain connected solely by non-covalent interactions.
S1/S2 での切断後、すべてのスパイクプロトマーは、非共有結合相互作用のみで接続されたままの 2 つのサブユニットで構成されます。
This event is suggested to convert the spike into its fusion-competent form as it could potentially promote the mobility of S1 receptor-binding domains (RBDs) and N-terminal domains (NTDs), resulting in decreased thermal stability, thereby facilitating the adaptation of the open conformation necessary for receptor binding.
このイベントは、スパイクを融合可能な形態に変換すると考えられています。これは、S1 受容体結合ドメイン (RBD) と N 末端ドメイン (NTD) の可動性を促進し、熱安定性を低下させ、受容体結合に必要なオープン構造の適応を促進する可能性があるためです。
Full cleavage at the S1/S2 interface is also assumed to be necessary for the first subunit to dissociate before refolding into the post-fusion state.
S1/S2 界面での完全な切断は、最初のサブユニットが解離して融合後の状態に再折り畳まれる前に必要であるとも考えられています。
3.2. Receptor recognition and binding
The receptor binding domains of the spike, which recognize and bind the ACE2 receptor, are located at the top of each spike in the S1 subunit.
スパイクの受容体結合ドメインは、ACE2 受容体を認識して結合し、S1 サブユニットの各スパイクの上部にあります。
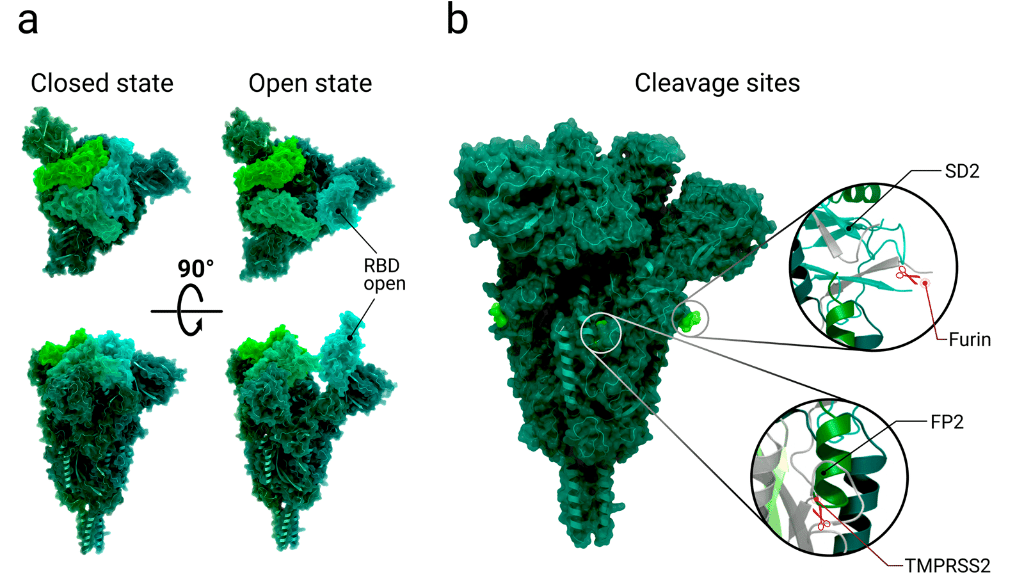
Each RBD (one per protomer) can adopt two conformations, such that it is oriented in the ‘up’ or ‘down’ position (Figure 2a).
各 RBD (プロトマーごとに 1 つ) は 2 つのコンフォメーションを採用でき、その結果、「上」または「下」の位置に配向されます (図 2a)。
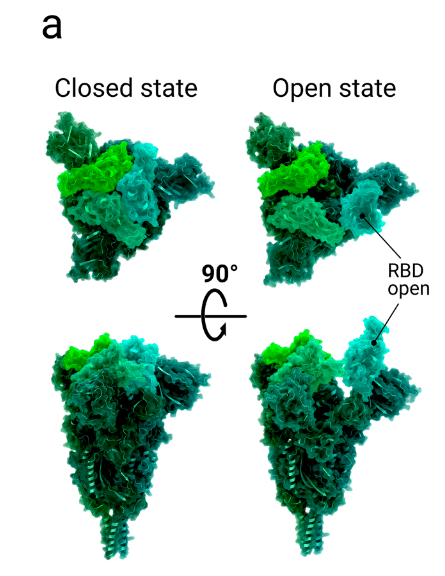
When the domain adopts the ‘up’ conformation, the receptor binding motif (RBM) is exposed and is available to interact with the surface cell receptor ACE2 or antibodies.
ドメインが「上」のコンフォメーションをとると、受容体結合モチーフ (RBM) が露出し、表面細胞受容体 ACE2 または抗体と相互作用できるようになります。
In its ‘down’ conformation, the RBD interacts with neither ACE2 nor antibodies.
RBD は「ダウン」コンフォメーションでは ACE2 とも抗体とも相互作用しません。
The dynamics of this domain are thus relevant for immune evasion.
このドメインのダイナミクスは免疫回避に関係しています。
An analysis of atomic B-factors supports flexibility or instability of the ‘down’ state of the RBD through increased disorder of the RBD compared to surrounding domains.
原子 B 因子の分析により、周囲のドメインと比較して RBD の無秩序性が増すことで、RBD の「ダウン」状態の柔軟性または不安定性が裏付けられます。
Structures of the receptor binding domain in complex with the human angiotensin converting enzyme 2 receptor reveal the interactions between the spike protein’s receptor binding motif and one of two binding-site lobes of the ACE2 N-terminal peptidase domain (c.f. PDB 7DQA, 6LZG, 6VW1).
ヒトアンジオテンシン変換酵素2受容体と複合体を形成した受容体結合ドメインの構造は、スパイクタンパク質の受容体結合モチーフとACE2 N末端ペプチダーゼドメインの2つの結合部位ローブの1つとの間の相互作用を明らかにしている(PDB 7DQA、6LZG、6VW1を参照)。
The contact between the receptor binding domain (with an interaction surface area of 864 Å2) and ACE2 (with an interaction surface area of 823 Å2) is established at the bottom side of the ACE2 small lobe, with a concave outer surface in the receptor binding motif accommodating the N-terminal helix of the ACE2, generating a total buried surface area of 1687 Å2 in total.
受容体結合ドメイン(相互作用表面積864Å2)とACE2(相互作用表面積823Å2)の接触は、ACE2小ローブの底部で確立され、受容体結合モチーフの凹状の外面がACE2のN末端ヘリックスに対応し、合計1687Å2の埋没表面積を生み出している。
Buried surface areas above approximately 700 Å2 indicate a rather tight binding which is stable in solution.
約 700 Å2 を超える埋没表面積は、溶液中で安定しているかなり密接な結合を示しています。
The RBM/RBD-ACE2 interface contains 13 hydrogen bonds and 2 salt bridges, with one salt-bridge outside of the RBM in the RBD between RBD Lys417 and ACE2 Asp30 (see Figure 2c).
RBM/RBD-ACE2 インターフェースには 13 個の水素結合と 2 個の塩橋が含まれており、RBD Lys417 と ACE2 Asp30 の間の RBD 内に RBM の外側に 1 個の塩橋があります (図 2c を参照)。
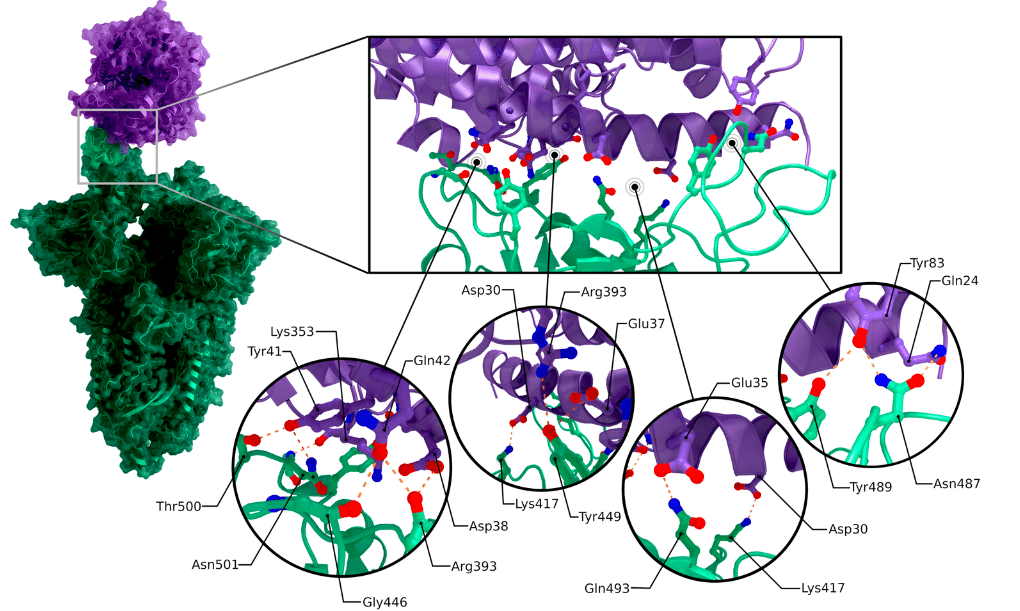
Even though the spike proteins of SARS-CoV-2 and SARS-CoV-1 share up to 76% sequence identity, their receptor binding motifs share only 47.8% sequence identity.
SARS-CoV-2 と SARS-CoV-1 のスパイクタンパク質は最大 76% の配列同一性を共有しているにもかかわらず、それらの受容体結合モチーフの配列同一性はわずか 47.8% です。
Mutation studies performed to evaluate how the sequence differences impact binding affinity to human ACE2 revealed that the substitution of amino acid Pro499, Gln493, Phe486, Ala475 and Leu455 in SARS-CoV-2 enhances ACE2 binding compared to SARS-CoV-1.
配列の違いがヒトACE2への結合親和性にどのように影響するかを評価するために実施された変異研究では、SARS-CoV-2のアミノ酸Pro499、Gln493、Phe486、Ala475、およびLeu455の置換により、SARS-CoV-1と比較してACE2結合が強化されることが明らかになりました。
Substitution of Gln493 to glutamate leads to substantial structural changes, removing an internal salt bridge between ACE2 residues Lys31 and Glu35 that is buried in a hydrophobic environment and replacing it by two hydrogen bonds between each of the residues to Glu493.
Gln493 をグルタミン酸に置換すると、構造的に大きな変化が生じ、疎水性環境に埋もれている ACE2 残基 Lys31 と Glu35 の間の内部塩橋が除去され、各残基と Glu493 の間の 2 つの水素結合に置き換えられます。
Lys31 of ACE2 forms another hydrogen bond with the non-polar Leu455.
ACE2 の Lys31 は、非極性の Leu455 と別の水素結合を形成します。
After binding the host cell surface receptor ACE2, domains of subunit S1 undergo several rearrangements.
宿主細胞表面受容体 ACE2 に結合した後、サブユニット S1 のドメインはいくつかの再配置を受けます。
One domain that is hypothesised to play a significant role within these rearrangements the fusion peptide proximal region (FPPR) (residues 828-853) which is situated next to SD1.
これらの再配置において重要な役割を果たすと仮定されているドメインの 1 つは、SD1 の隣にある融合ペプチド近位領域 (FPPR) (残基 828-853) です。
The structure PDB 6XR8 with its RBD in ‘down’ conformation shows the FPPR in an ordered state, packed around an internal disulphide bond between cysteines C840 and C851.
RBD が「下向き」のコンフォメーションにある構造 PDB 6XR8 は、システイン C840 と C851 の間の内部ジスルフィド結合の周りに詰め込まれた、整然とした状態の FPPR を示しています。
This packing is reinforced by an extensive hydrogen network and an additional salt bridge between Lys835 and Asp848.
この詰め込みは、広範な水素ネットワークと Lys835 と Asp848 の間の追加の塩橋によって強化されています。
Superimposing the S2 domain of 6XR8, which contains the ordered FPPR, with a structure where the receptor binding domain is in the ‘up’ conformation (PDB 6VSB [Citation64]) results in clashes between the FPPR and an outwards-rotated sub-domain 1 (SD1; Figure 3a and 3b).
整列した FPPR を含む 6XR8 の S2 ドメインを、受容体結合ドメインが「上向き」の構造 (PDB 6VSB [引用 64]) に重ねると、FPPR と外側に回転したサブドメイン 1 (SD1、図 3a および 3b) が衝突します。
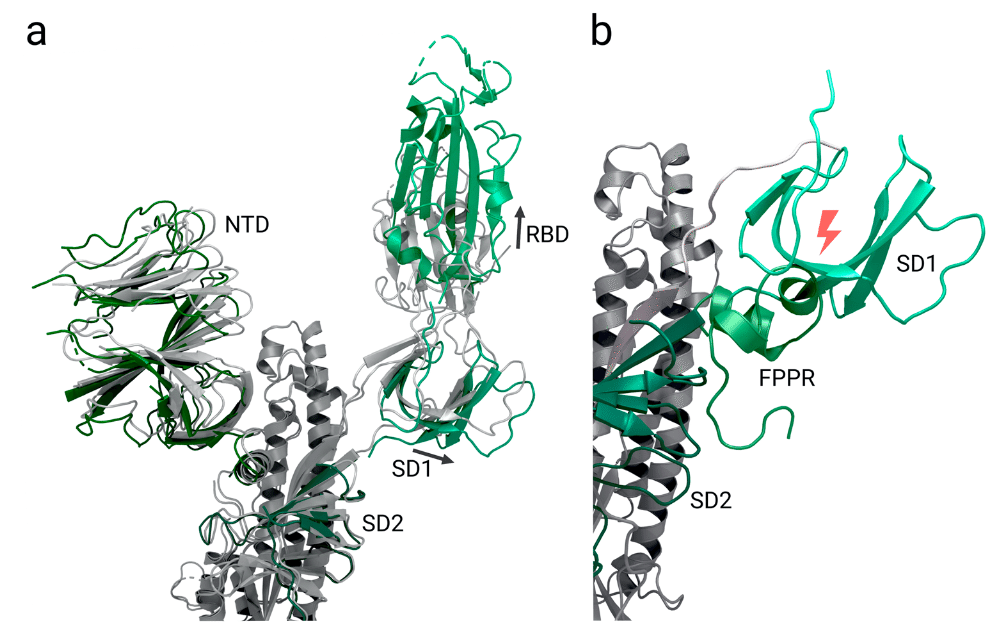
Thus, it is speculated that in the ‘up’ conformation, the FPPR is delocalized whereas in the closed state it helps to clamp down the receptor binding domain and stabilizes the ‘down’ conformation.
したがって、「上」のコンフォメーションでは FPPR は非局在化されるのに対し、閉じた状態では受容体結合ドメインを締め付けて「下」のコンフォメーションを安定化させると推測されます。
SD1 is supposed to act as a relay between both domains (Figure 3b).
SD1 は両ドメイン間の中継役として機能すると考えられています (図 3b)。
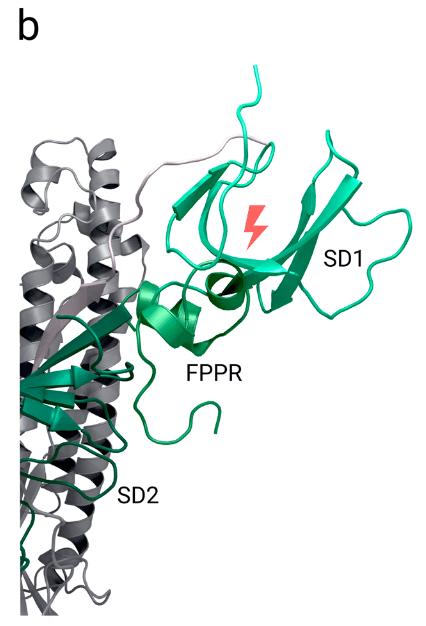
A proposed mechanism suggests that the stochastic thermal departure of FPPR from its stabilizing position enables the SD1 to rotate and the receptor binding domain to change to the up conformation to engage ACE2.
提案されたメカニズムは、FPPR が安定位置から熱的に離脱することで SD1 が回転し、受容体結合ドメインが上向きの構造に変化して ACE2 と結合できるようになることを示唆しています。
The ‘up’ conformation is then fixed when the receptor binding motif binds the ACE2 receptor of the host cell, thereby leading to exposure of the S2’ cleavage site upstream of the fusion peptide.
受容体結合モチーフが宿主細胞の ACE2 受容体に結合すると、「上向き」コンフォメーションが固定され、融合ペプチドの上流の S2 切断部位が露出します。
It's important to note that this proposed mechanism is based on theoretical considerations and requires further investigation for validation.
ここで重要なのは、この提案されたメカニズムは理論的な考察に基づいており、検証にはさらなる調査が必要であるということです。
By comparing the native spike with one receptor binding domain in the ‘up’ position while either unbound or bound to ACE2, the binding to ACE2 appears to shift the position of the extended RBD, amounting to a rigid-body rotation that moves the RBD’s centre of mass approximately 5.5 Å away from the trimer axis.
ネイティブスパイクを、結合していないか ACE2 に結合している状態で「上」の位置にある 1 つの受容体結合ドメインと比較すると、ACE2 への結合により、拡張された RBD の位置がシフトするように見えます。 これは、RBD の質量中心を三量体軸から約 5.5 Å 離す剛体回転に相当します。
Additionally, the subdomains associated with the RBD (SD1) as well as the N-terminal domain (SD2) of the same monomer translate ∼1.9 Å and ∼2.3 Å, respectively.
さらに、同じモノマーの RBD (SD1) に関連するサブドメインと N 末端ドメイン (SD2) は、それぞれ約 1.9 Å と約 2.3 Å 移動します。
Within this translation, all three N-terminal domains move by roughly 1.5-3.0 Å.
この移動では、3 つの N 末端ドメインすべてが約 1.5~3.0 Å 移動します。
The flexibility of spikes has been proposed to promote receptor recognition.
スパイクの柔軟性は受容体の認識を促進すると考えられている。
Cryo-electron tomography shows that spikes are highly variable with a standard deviation of 20° about the average deviation of 40° away from a perpendicular orientation to the viral membrane.
クライオ電子トモグラフィーでは、スパイクはウイルス膜に対する垂直方向から平均偏差 40° 離れた標準偏差 20° で非常に変動性が高いことが示されています。
The sparse distribution of spikes on the viral envelope of SARS-CoV-2 particles may be crucial for allowing this flexibility, a characteristic that is markedly distinct to other class I fusion proteins.
SARS-CoV-2 粒子のウイルスエンベロープ上のスパイクのまばらな分布は、この柔軟性を可能にするために重要であり、他のクラス I 融合タンパク質とは著しく異なる特徴である。
Several Y-shaped spike assemblies were also observed on the viral surface: due to the high degree of rotational freedom, adjacent spikes had intertwined their stems.
ウイルス表面には、Y 字型のスパイク集合体もいくつか観察されました。回転の自由度が高いため、隣接するスパイクは茎を絡み合わせていました。
This feature may allow the spikes to better engage ACE2 receptors but a sparsely occupied surface may also make these molecules more vulnerable to antibody recognition.
この特徴により、スパイクは ACE2 受容体とよりよく結合できる可能性があるが、まばらに占有された表面はこれらの分子を抗体認識に対してより脆弱にする可能性がある。
3.3. Post-binding spike protein cleavage and activation
結合後のスパイクタンパク質の切断と活性化
For the cleavage event that follows binding to the receptor, two different proteases are currently under consideration, depending on the viral cell entry route.
受容体への結合後に起こる切断イベントについては、ウイルスの細胞侵入経路に応じて、現在 2 つの異なるプロテアーゼが検討されています。
Viral cell entry was observed to occur either through the plasma membrane pathway via the cell surface or the endosomal pathway after virus endocytosis.
ウイルス細胞侵入は、細胞表面を介した細胞膜経路またはウイルスエンドサイトーシス後のエンドソーム経路のいずれかで発生することが観察されました。
In the plasma membrane pathway, type II transmembrane serine protease (TMPRSS2) is thought to have a significant role by cleaving a monobasic site at location S2’ (Table 3), possibly being necessary for releasing the fusion peptides.
細胞膜経路では、II型膜貫通セリンプロテアーゼ(TMPRSS2)がS2’(表3)の単塩基部位を切断することで重要な役割を果たしていると考えられており、融合ペプチドの放出に必要である可能性があります。
This cell surface protein is widely expressed in epithelial cells of the respiratory and gastrointestinal tract.
この細胞表面タンパク質は、呼吸器および消化管の上皮細胞で広く発現しています。
In studies where target cells lacked cell surface proteases like TMPRSS2, SARS-CoV-2 was internalized via endocytosis, with spike protein cleavage likely to be executed by cathepsin B or L.
標的細胞に TMPRSS2 のような細胞表面プロテアーゼが存在しない研究では、SARS-CoV-2 はエンドサイトーシスによって内部化され、スパイクタンパク質の切断はカテプシン B または L によって実行される可能性が高い。
The exact cleavage site/sites remain in discussion. Proposed locations are either directly at S2’ or upstream of this site.
正確な切断部位については議論が続いています。提案されている場所は、S2’ のすぐ上か、この部位の上流です。
What cause (e.g. spike protein cleavage, dissociation of S1 or others) triggers the rearrangements from the prefusion to the post-fusion state is not fully clear and remains to be investigated.
融合前状態から融合後状態への再配置を引き起こす原因(スパイクタンパク質の切断、S1 の解離など)は完全には解明されておらず、調査が必要です。
The exact mechanism of the pre- to post-fusion transition of SARS-CoV-2 as well as membrane fusion itself is not yet fully understood.
SARS-CoV-2 の融合前状態から融合後状態への移行の正確なメカニズムと膜融合自体は、まだ完全には解明されていません。
However, based on research of different fusion proteins including SARS-CoV-2 and 1, mechanisms for the rearrangements in the spike protein’s subunit S2 and the membrane fusion are proposed in the following three sections.
ただし、SARS-CoV-2 および 1 を含むさまざまな融合タンパク質の研究に基づいて、スパイクタンパク質のサブユニット S2 の再配置と膜融合のメカニズムが次の 3 つのセクションで提案されています。
Hypothetical rearrangement in S2 to form the fusion peptide ‘complex’
S2 における仮説的な再配置により融合ペプチド「複合体」が形成される
Formation of the fusion peptide ‘complex’ after shedding of S1 entails the conformational rearrangements of the heptad repeat 1 (HR1) and heptad repeat 2 (HR2) domains, exposing the proposed fusion peptide regions (FP1 and FP2), which subsequently insert into the host cell membrane.
S1 の切断後の融合ペプチド「複合体」の形成には、ヘプタド リピート 1 (HR1) ドメインとヘプタド リピート 2 (HR2) ドメインの立体配座の再配置が伴い、提案された融合ペプチド領域 (FP1 と FP2) が露出し、その後、宿主細胞膜に挿入されます。
As a first step in the formation of the complex, heptad repeat 1 rearranges to form a 180 Å-long three-stranded coiled-coil together with the central helix (CH) domain.
複合体形成の最初のステップとして、ヘプタドリピート 1 は再配置され、中央ヘリックス (CH) ドメインとともに 180 Å の長さの 3 本鎖コイルドコイルを形成します。
Simultaneously a section of heptad repeat 2 (I1169 to Q1180) adopts an extended structure with the remaining part of heptad repeat 2 forming a canonical α-helical structure.
同時に、ヘプタドリピート2のセクション(I1169からQ1180)は拡張構造を採用し、ヘプタドリピート2の残りの部分は標準的なαヘリックス構造を形成します。
It is suggested that, similar to proposed mechanisms of other coronaviruses, the extension of the long central helix by inclusion of the N-terminal section of heptad repeat 1 (amino acids 990–1035) projects the fusion peptide in the direction of the host cell membrane.
他のコロナウイルスの提案されたメカニズムと同様に、ヘプタッドリピート1のN末端セクション(アミノ酸990〜1035)を含めることによって長い中央ヘリックスが延長され、融合ペプチドが宿主細胞膜の方向に突き出ると示唆されています。
The dissociation of S1 and cleavage of the S2´ site at 815R|S816 by type II transmembrane serine protease (TMPRSS2) is suggested to release the fusion peptides that contain several conserved amino acids as the new N-terminus of S2.
S1 の解離と、タイプ II 膜貫通セリンプロテアーゼ (TMPRSS2) による 815R|S816 の S2´ 部位の切断により、S2 の新しい N 末端としていくつかの保存されたアミノ酸を含む融合ペプチドが放出されると示唆されています。
Located on the first consecutive section (FP1), there is an additional conserved hydrophobic LLF motif (L821/L822/F823) which is thought to penetrate the target cell membrane and was previously shown to be crucial for interaction of the SARS-CoV-1 fusion peptides with lipid membranes.
最初の連続セクション (FP1) には、保存された疎水性 LLF モチーフ (L821/L822/F823) がさらに存在し、これは標的細胞膜を貫通すると考えられており、SARS-CoV-1 融合ペプチドと脂質膜の相互作用に重要であることが以前に示されています。
The released fusion peptides trimerize to form a so-called ‘spearhead’ that anchors the spike in the target cell membrane through its hydrophobic residues.
放出された融合ペプチドは三量体化して、いわゆる「槍先」を形成し、その疎水性残基を介して標的細胞膜にスパイクを固定します。
FP1 and FP2 are suggested to form a bipartite membrane interaction platform where certain residues enhance binding of the host cell membrane by interaction with Ca2+ ions.
FP1 と FP2 は、特定の残基が Ca2+ イオンとの相互作用によって宿主細胞膜の結合を強化する二分膜相互作用プラットフォームを形成することが示唆されています。
At the same time, the cysteine rich CT that anchors the C-terminal part of the spike in the viral membrane gets reinforced by a membrane-actin linker named ezrin.
同時に、ウイルス膜のスパイクの C 末端部分を固定するシステインに富む CT は、エズリンと呼ばれる膜アクチンリンカーによって強化されます。
After conformational rearrangements in S2, the membranes are in close apposition for fusion.
S2 での構造再配置後、膜は融合のために近接する位置にある。
Rearrangements in S2 leading to fusion and formation of post-fusion hairpin state.
S2 の再配置により融合が起こり、融合後のヘアピン状態が形成されます。
Based on studies of other Class I virus fusion proteins it has been proposed that after inserting the spearhead into the host cell membrane, the two membranes are pulled together to initiate membrane fusion.
他のクラス I ウイルス融合タンパク質の研究に基づいて、スピアヘッドが宿主細胞膜に挿入された後、2 つの膜が引き寄せられて膜融合が開始されると考えられています。
As a result of this movement, the spike refolds into a closing hairpin structure.
この動きの結果、スパイクは閉じたヘアピン構造に折り畳まれます。
Here, heptad repeats 1 and 2 play a crucial part: The repeated hydrophobic amino acids from the HPPHCPC motifs form a hydrophobic interface and drive heptad-repeat hairpin trimerization and furthermore six-helix bundle formation for membrane fusion with the host cell.
ここで、ヘプタドリピート 1 と 2 が重要な役割を果たします。HPPHCPC モチーフからの繰り返し疎水性アミノ酸は疎水性インターフェースを形成し、ヘプタドリピートヘアピン三量体化を促進し、さらに宿主細胞との膜融合のための 6 ヘリックスバンドルの形成を促進します。
The post-fusion state of fusion domains, in particular the six-helix bundle or post-fusion core, has also been relatively well characterized through research of various viruses such as HIV gp41, parainfluenza virus F, murine hepatitis virus, and SARS-CoV.
融合ドメインの融合後状態、特に6ヘリックス束または融合後コアは、HIV gp41、パラインフルエンザウイルスF、マウス肝炎ウイルス、SARS-CoVなどのさまざまなウイルスの研究を通じて、比較的よく特徴付けられています。
In the first post-fusion bundle of six helices, the hydrophobic groove of HR1 is bound in an antiparallel manner by the N-terminal section of HR2, which adopts a helical conformation with one turn.
融合後の最初の 6 本のヘリックス束では、HR1 の疎水性溝が HR2 の N 末端部分によって反平行に結合され、1 回転のヘリックス構造をとっています。
Together with the C-terminal domain of HR2, this organizes into a longer helix; the remaining part of the coiled coil of HR1 arranges in another bundle of six helices (see Figure 4a and 4b).
HR2 の C 末端ドメインと一緒に、これはより長いヘリックスに組織化されます;HR1 のコイルドコイルの残りの部分は、6 つのヘリックスの別の束に配置されます (図 4a および 4b を参照)。
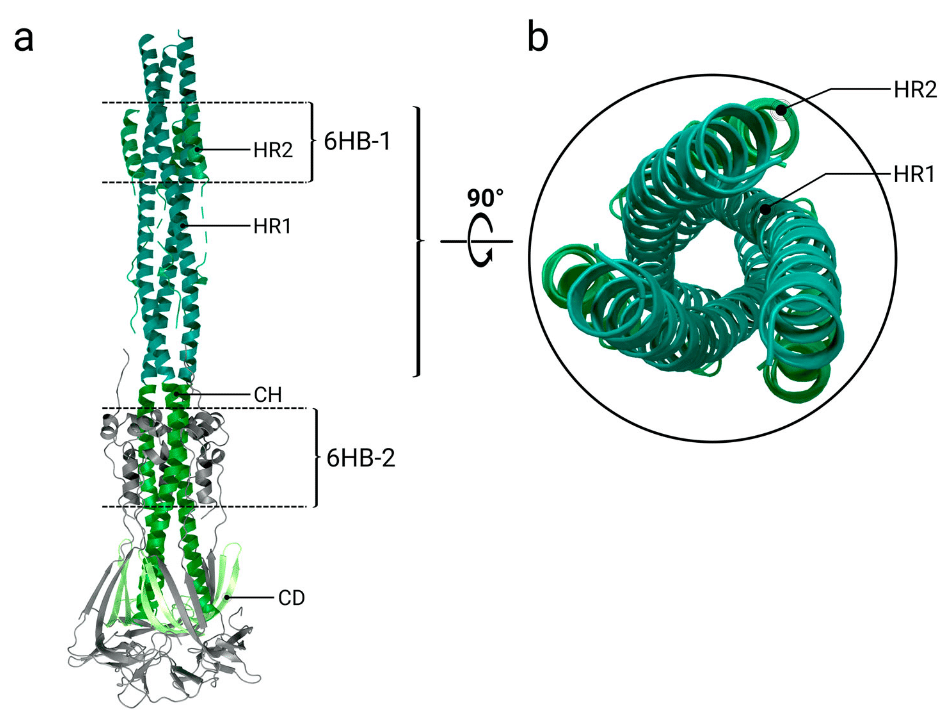
Structural analysis of interactions between both HR domains reveals that, as well as hydrophobic interactions, multiple hydrogen bonds between the two domains occur (Asn925, Gln935, Gln949, Asn953, Asn960 of HR1 and Ala1174, Val1177, Ile1179, Gln1180, Ala1190, Asn1194, Ile1198 of HR2).
両方の HR ドメイン間の相互作用の構造分析により、疎水性相互作用だけでなく、2 つのドメイン間で複数の水素結合が発生することが明らかになりました (HR1 の Asn925、Gln935、Gln949、Asn953、Asn960、および HR2 の Ala1174、Val1177、Ile1179、Gln1180、Ala1190、Asn1194、Ile1198)。
These multiple reinforcements of the central coiled coils result in a stable post-fusion structure.
中央のコイルドコイルのこれらの多重強化により、安定した融合後構造が生まれます。
In the prefusion state, the connector domain folds collectively with a section of the S1/S2´ fragment (amino acid 718-729) into a three stranded β-sheet.
融合前の状態では、コネクタドメインは S1/S2´ フラグメントの一部 (アミノ酸 718-729) と集合的に折り畳まれ、3 本の鎖からなる β シートを形成します。
During the transition to the post-fusion state, the β-sheet does not alter position but instead is expanded by another strand formed of residues 1127–1135 (between connector domain and heptad repeat 2), as the C-terminal HR2 domain is projected towards the viral membrane.
融合後状態への移行中、βシートの位置は変化せず、代わりに残基1127~1135(コネクタドメインとヘプタッドリピート2の間)で形成された別の鎖によって拡張され、C末端HR2ドメインがウイルス膜に向かって突出します。
Amino acids 737–769 from each spike protomer form a helical region which packs against the grooves of the CH forming another six-helix-bundle structure locked in place through disulphide bonds.
各スパイクプロトマーのアミノ酸737~769はらせん領域を形成し、これがCHの溝に詰め込まれ、ジスルフィド結合によって固定された別の6つのらせん束構造を形成します。
Both of these fragments are an integral part of the post fusion structure, and do not dissociate even after the cleavage event at the S2´ site.
これら両方の断片は融合後の構造の不可欠な部分であり、S2´部位での切断イベント後でも解離しません。
3.4. Membrane fusion mediated by the fusion peptide
融合ペプチドによる膜融合
After insertion of the fusion peptide (FP) into the host cell membrane, and the formation of the hairpin post-fusion structure, both membranes are brought into close proximity.
融合ペプチド (FP) が宿主細胞膜に挿入され、融合後のヘアピン構造が形成されると、両方の膜が近接します。
The FP then mediates viral and host cell membrane fusion by disrupting and connecting lipid bilayers.
その後、FP は脂質二重層を破壊して結合させることで、ウイルスと宿主細胞の膜融合を仲介します。
During the fusion process, a fusion pore is formed enabling the transfer of the viral genetic material into the host cell.
融合プロセス中に、融合孔が形成され、ウイルスの遺伝物質を宿主細胞に輸送できるようになります。
To date, there is no molecular-level data about the structure of this spearhead or its interaction with the cell membrane and the mechanism leading to formation of the fusion pore is only poorly understood.
現在までに、このスピアヘッドの構造や細胞膜との相互作用に関する分子レベルのデータはなく、融合孔の形成につながるメカニズムはほとんど解明されていない。
Other regions which might also contribute to the fusion process have also not yet been sufficiently investigated, such as a conserved hydrophobic region next to the transmembrane domain that is necessary for trimerization of the spike protein and the connector domain region.
融合プロセスに寄与する可能性のある他の領域も、スパイクタンパク質の三量体化に必要な膜貫通ドメインの隣の保存された疎水性領域とコネクタドメイン領域など、まだ十分に調査されていません。
[Citation18]. Hence, a series of questions regarding the fusion process remain unanswered.
したがって、融合プロセスに関する一連の疑問は未解決のままである。
A schematic illustration of the fusion process is given in Figure 4c.
融合プロセスの概略図を図4cに示す。
4. Glycosylation
グリコシル化
Spikes of enveloped viruses are heavily glycosylated.
エンベロープウイルスのスパイクは高度にグリコシル化されています。
Viral glycosylation plays a major role in many different intrinsic and extrinsic properties of the complex, being essential to folding, trafficking and regulating interactions with lectins while also modulating binding to other receptors and glycans that mediate host cell entry.
ウイルスのグリコシル化は、複合体のさまざまな内因性および外因性特性において重要な役割を果たしており、折り畳み、輸送、レクチンとの相互作用の調節に不可欠であると同時に、宿主細胞への侵入を媒介する他の受容体やグリカンへの結合も調節します。
The characteristic dense coat of complex carbohydrates, also known as a glycan shield, retains a high degree of flexibility, dynamics and microheterogeneity.
複合炭水化物の特徴的な高密度コートはグリカンシールドとも呼ばれ、高度な柔軟性、ダイナミクス、および微小不均一性を維持しています。
In addition to these functions, the highly dense glycan shield is known to effectively mask the virus from the immune system, protecting viral protein from recognition by cells and antibodies.
これらの機能に加えて、高密度グリカンシールドは、ウイルスを免疫システムから効果的に隠蔽し、細胞や抗体によるウイルスタンパク質の認識を防ぐことが知られています。
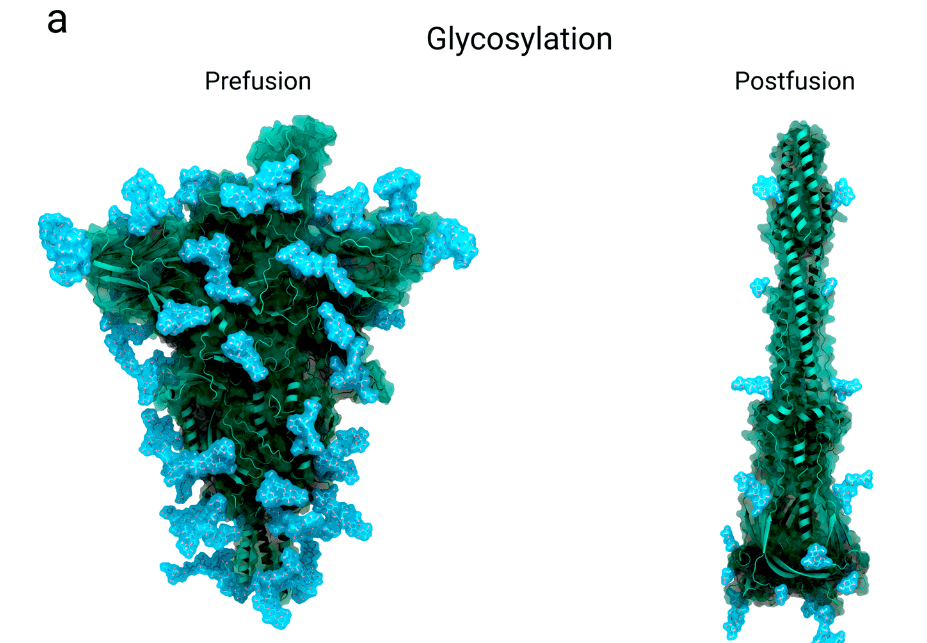
Each SARS-CoV-2 spike protomer contains 22 N-glycosylation sites (sequons).
各 SARS-CoV-2 スパイクプロトマーには、22 個の N グリコシル化部位 (セクオン) が含まれています。
Of these, 18 are consistently occupied, with predominantly complex N-glycan structures, and likely shield more than 60% of the surface of a spike’s head (S1) from antibody-sized molecules (i.e. scFVs, mAbs, nanobodies, etc; Figure 5a).
これらのうち 18 個は一貫して占有されており、主に複雑な N 型糖鎖構造を持ち、スパイクの頭部 (S1) の表面の 60% 以上を抗体サイズの分子 (scFV、mAbs、ナノボディなど、図 5a) から保護していると考えられます。
For smaller molecules, the shielding is suggested to be less effective.
より小さな分子の場合、遮蔽の効果は低いと示唆されている。
17 O-glycosylation sites have also been identified, with a much lower degree of occupancy, averaging less than 2%.
17 個の O-グリコシル化部位も特定されていますが、占有率ははるかに低く、平均 2% 未満です。
Of these 17 glycosylation sites, 11 are located on subunit S1 and the remaining 6 on subunit S2.
これらの 17 個のグリコシル化部位のうち、11 個はサブユニット S1 上に、残りの 6 個はサブユニット S2 上にあります。
An essential aspect of this molecular disguise is that the spike protein is glycosylated by the host cell machinery, mimicking the natural glycosylation of the host’s proteins.
この分子偽装の重要な側面は、スパイクタンパク質が宿主細胞の機構によってグリコシル化され、宿主タンパク質の自然なグリコシル化を模倣することです。
The spike protein’s N-glycans are linked to the protein surface in the endoplasmic reticulum and further refined in the Golgi apparatus of the infected host cell.
スパイクタンパク質の N-グリカンは小胞体のタンパク質表面に結合し、感染した宿主細胞のゴルジ体でさらに精製されます。
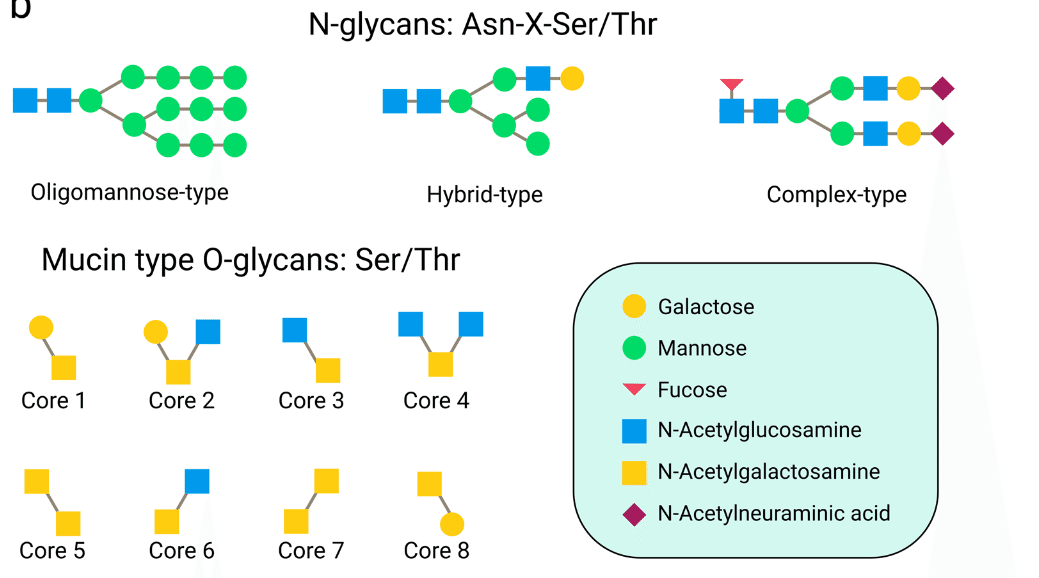
While the viruses move through the Golgi lumina, N-glycans undergo extensive processing by glycosylation enzymes. O-glycosylation also occurs in the Golgi.
ウイルスがゴルジ体腔を通過する間、N-グリカンはグリコシル化酵素によって広範囲に処理されます。ゴルジ体ではO-グリコシル化も起こります。
An overview over the types of N-glycans and O-glycans functionalizing the spike is given in Figure 5b.
スパイクを機能化する N-グリカンと O-グリカンの種類の概要を図 5b に示します。
The host protease furin, or some similar enzyme, cleaves the spike protomer at the S1/S2 interface before the virus gets secreted via lysosomes into the extracellular surroundings, and O-glycosylation has been shown to have both positive and negative impacts on proteolytic cleavage events.
宿主プロテアーゼであるフーリンまたは類似の酵素は、ウイルスがリソソームを介して細胞外環境に分泌される前に、S1/S2 界面でスパイクプロトマーを切断します。O-グリコシル化は、タンパク質分解切断イベントにプラスとマイナスの両方の影響を与えることが示されています。
Therefore, differences in O-glycan expression may potentially modulate this protease activity and thus viral infectivity.
したがって、O-グリカン発現の違いは、このプロテアーゼ活性、ひいてはウイルスの感染性を潜在的に調節する可能性があります。
The heterogeneous nature of the glycan shield also precludes experimental structural characterization.
グリカンシールドの不均一な性質は、実験による構造特性評価も妨げます。
Thus, most structural studies of the viral glycan shield rely on molecular dynamics simulations.
したがって、ウイルスのグリカンシールドの構造研究のほとんどは、分子動力学シミュレーションに依存しています。
All-atom molecular simulations are highly complementary to glycoproteomics studies that indicate the type of glycan structures and the relative occupancy of the sites of glycosylation, and to cryo-EM data.
全原子分子シミュレーションは、グリカン構造の種類とグリコシル化部位の相対的占有率を示すグリコプロテオミクス研究、およびクライオ電子顕微鏡データと非常に補完的です。
One limitation is that with cryo-EM only the most rigid (and commonly shared) part of the glycan structure can be resolved as shown in the post-fusion state in Figure 5a.
1 つの制限は、クライオ電子顕微鏡では、図 5a の融合後状態に示すように、グリカン構造の最も硬い (そして一般的に共有されている) 部分しか解析できないことです。
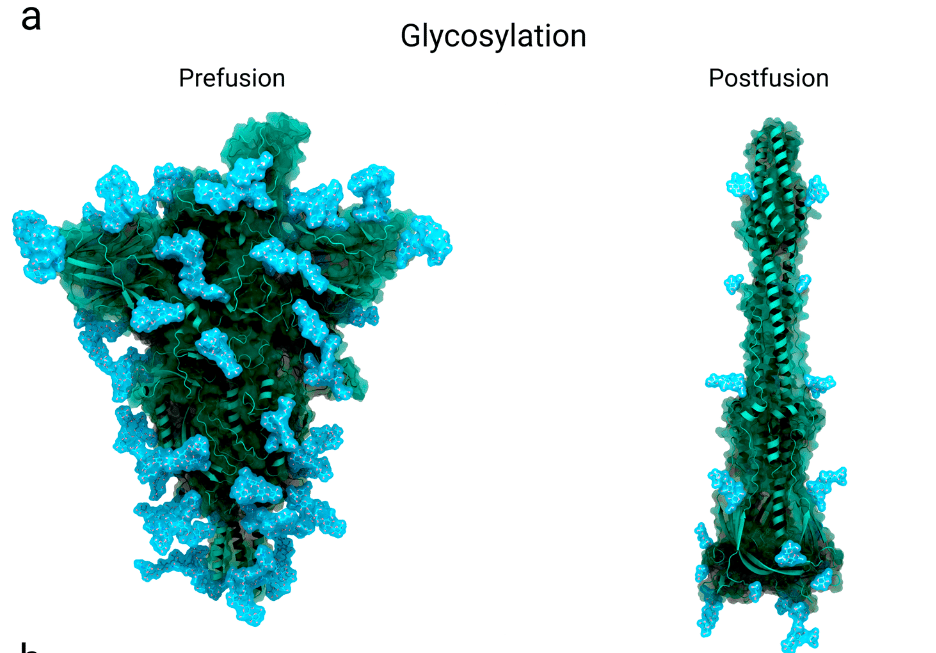
As a unique feature of the SARS-CoV-2 spike, the glycan shield is also mechanistically functional.
SARS-CoV-2 スパイクのユニークな特徴として、グリカン シールドはメカニズム的にも機能します。
Three important N-glycan sites, Asn165, Asn343 and Asn234 mediate the RBD conformational dynamics with a focus on stabilizing the RBD ‘up’ conformation by a ‘load and lock’ mechanism.
3 つの重要な N グリカン サイト、Asn165、Asn343、および Asn234 は、RBD の「アップ」コンフォメーションを「ロード アンド ロック」メカニズムによって安定化することに重点を置いて、RBD コンフォメーション ダイナミクスを媒介します。
The mutations N165A/N234A and N234A lead to a significantly reduced ACE2 binding and cause a shift in the RBD conformational ensemble towards the ‘down’ conformation.
変異 N165A/N234A および N234A は ACE2 結合を大幅に減少させ、RBD 構造集合体を「ダウン」構造へとシフトさせます。
5. Therapeutic interest
治療への関心
5.1. Antigenic epitopes in natural immune responses and vaccine development
自然免疫反応とワクチン開発における抗原エピトープ
As the largest protein on the viral surface, the spike protein is the main antigen targeted by the immune system, particularly the receptor binding domain.
ウイルス表面で最も大きなタンパク質であるスパイクタンパク質は、免疫系、特に受容体結合ドメインが標的とする主な抗原です。
The spike can provoke both a strong T-cell response and a neutralizing immune response, inducing protective immunity against COVID-19.
スパイクは強力なT細胞反応と中和免疫反応の両方を引き起こし、COVID-19に対する防御免疫を誘導します。
This makes the surface protein attractive both as a target for development of vaccines and for drugs against SARS-CoV-2.
このため、表面タンパク質はワクチン開発のターゲットとしても、SARS-CoV-2に対する薬剤のターゲットとしても魅力的です。
One study of infected patients showed that monoclonal antibodies raised against the spike distributed reasonably well across the different regions.
感染患者を対象としたある研究では、スパイクに対して生成されたモノクローナル抗体がさまざまな領域にかなりよく分布していることが示されました。
Of 377 antibodies isolated from 77 recovered COVID-19 patients, 34% bind to S1 and 53% bind to S2.
回復した 77 人の COVID-19 患者から分離された 377 個の抗体のうち、34% が S1 に結合し、53% が S2 に結合しました。
The remaining 13% interact with none of the subdomains.
残りの13%は、どのサブドメインとも相互作用しません。
Of the antibodies that bind to S1, 21% bind to the RBD and 11% to the NTD.
S1に結合する抗体のうち、21%がRBDに、11%がNTDに結合します。
Almost all inherently neutralising antibodies bind to the RBD, along with a smaller set which bind to an ‘antigenic supersite’ on the N-terminal domain located at the most radially distant loops from the centre of the spike.
本質的に中和する抗体のほぼすべてが RBD に結合し、スパイクの中心から最も放射状に離れたループに位置する N 末端ドメインの「抗原性スーパーサイト」に結合するより小さなセットも結合します。
However, one class of antibodies, originally discovered through cross-reactivity from SARS-CoV-1 antibody CR3022 and then rediscovered in patient-derived antibodies as EY6A, achieves neutralization by forcing RBDs to an ‘up and out’ conformation which shows destabilization of the spike protein in cryo-EM images.
しかし、もともとSARS-CoV-1抗体CR3022との交差反応を通じて発見され、その後EY6Aとして患者由来の抗体で再発見されたあるクラスの抗体は、RBDを「上向き外向き」の構造に強制することで中和を達成します。 これは、クライオ電子顕微鏡画像でスパイクタンパク質の不安定化を示しています。
The immunodominance of the receptor binding domain is accounted for by the extensive shielding of most spike exodomains by N- or O-linked glycans.
受容体結合ドメインの免疫優位性は、ほとんどのスパイク外ドメインがN結合型またはO結合型グリカンによって広範囲に遮蔽されていることで説明されます。
Additionally, as it mediates specificity in cell tropism, it elicits highly functionally relevant antibodies which prevent RBD-ACE2 interaction while avoiding the risk of antibody-dependent enhancement that could be mediated by weak or non-neutralizing antibodies.
さらに、細胞向性の特異性を媒介するため、RBD-ACE2 相互作用を阻止する機能的に非常に関連のある抗体を誘導し、弱い抗体や非中和抗体によって媒介される可能性のある抗体依存性増強のリスクを回避します。
Rather than contributing to the immunity, such non-neutralizing antibodies facilitate viral entry and subsequent infection of the host cell, leading to an increased virulence.
このような非中和抗体は免疫に寄与するのではなく、ウイルスの侵入とそれに続く宿主細胞への感染を促進します。 その結果、毒性が高まります。
One example of such an epitope in SARS-CoV-1 is found nearby to the RBD: antibodies targeting this region markedly enhance the effectiveness infection.
SARS-CoV-1 のそのようなエピトープの一例は、RBD の近くに見つかります:この領域を標的とする抗体は、感染の有効性を著しく高めます。
The RBD also contains epitopes for T-cell response.
RBD には、T 細胞応答のエピトープも含まれています。
Most of the currently developed vaccines, encompassing both vector-based as well as mRNA-based vaccines, include the RBD in their immunogens.
現在開発されているワクチンのほとんどは、ベクターベースとmRNAベースのワクチンの両方を含み、免疫原にRBDが含まれています。
Other epitopes on the spike targeted by neutralizing antibodies are found on the NTD of the S1 subunit.
中和抗体が標的とするスパイク上の他のエピトープは、S1 サブユニットの NTD にあります。
Antibodies targeting these regions do not directly prevent receptor binding but rather interfere with it or restrain the rearrangements from the prefusion to the post-fusion conformation.
これらの領域を標的とする抗体は、受容体の結合を直接阻止するのではなく、むしろそれを妨害するか、融合前構造から融合後構造への再配置を抑制します。
These antibodies show weaker neutralizing potency than those targeting the RBD.
これらの抗体は、RBD を標的とする抗体よりも中和効力が弱い。
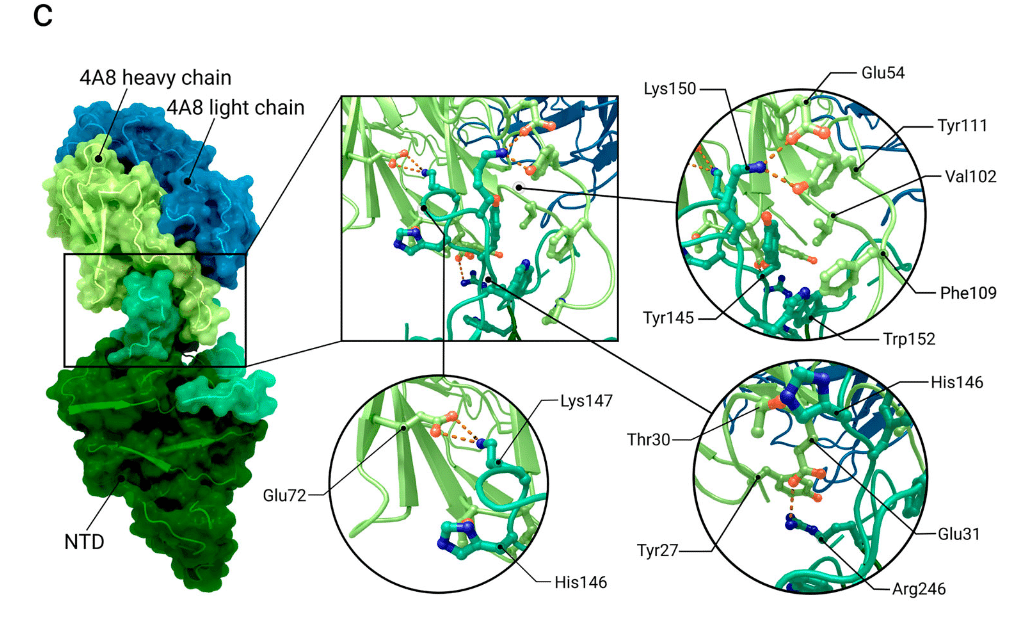
Structural analysis of antibodies in complex with the N-terminal domain (NTD) revealed that the interaction takes place primarily within two glycan-free surfaces named the NTD-1 and NTD-2 regions (Figure 6a).
N末端ドメイン(NTD)と複合体を形成した抗体の構造解析により、相互作用は主にNTD-1およびNTD-2領域と呼ばれる2つのグリカンフリー表面内で起こることが明らかになりました(図6a)。
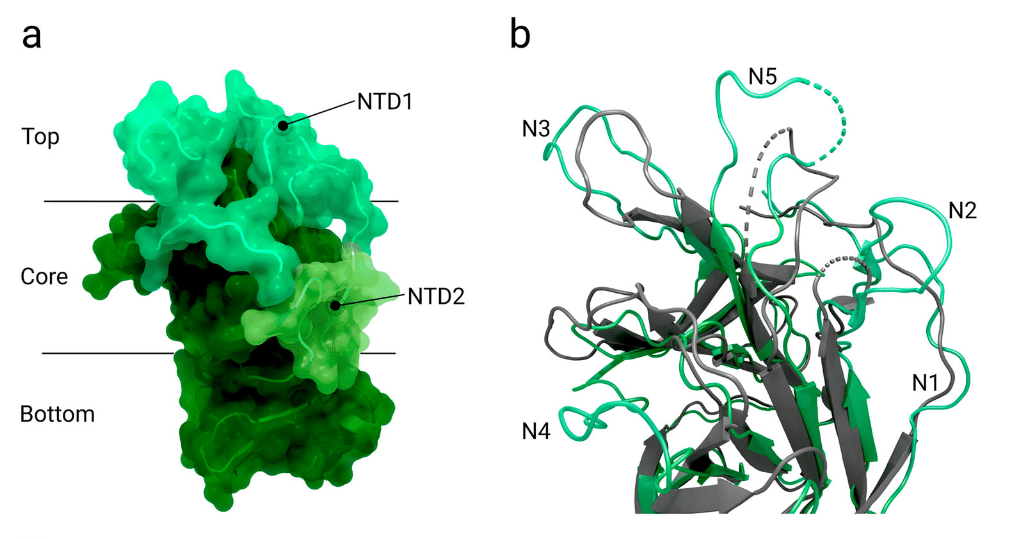
NTD-1, located in the NTD top-core region, is targeted by most antibodies and is designated as the NTD-1-antigenic supersite.
NTD-1 は NTD トップコア領域に位置し、ほとんどの抗体の標的となり、NTD-1 抗原スーパーサイトとして指定されています。
This site comprises five loop structures named N1-5 as well as an additional β-hairpin structure near N3.
この部位は、N1-5 と名付けられた 5 つのループ構造と、N3 付近の追加の β ヘアピン構造で構成されています。
The loop structures span the amino acids 14–26 (N1), 67–79 (N2), 141–156 (N3), 177–186 (N4) and 246–260 (N5).
ループ構造は、アミノ酸 14~26 (N1)、67~79 (N2)、141~156 (N3)、177~186 (N4)、および 246~260 (N5) にまたがっています。
Different reconfigurations of the loops are observed for different antibodies binding to the highly conserved NTD-1 supersite (Figure 6b and 6c).
高度に保存された NTD-1 スーパーサイトに結合する抗体によって、ループの異なる再構成が観察されます (図 6b および 6c)。
The part of subunit S2 proximal to the viral membrane also poses a potential target for neutralizing antibodies, even though it is more extensively shielded by glycans, making it less immunogenic.
ウイルス膜に近いサブユニット S2 の部分も、グリカンによってより広範囲に保護されているため免疫原性が低いものの、中和抗体の潜在的な標的となる。
It is possible that antibodies targeting S2 might interfere with the structural rearrangement of the spike protein and the insertion of the FP required for virus–host membrane fusion.
S2を標的とする抗体は、スパイクタンパク質の構造的再配置と、ウイルスと宿主の膜融合に必要なFPの挿入を妨害する可能性がある。
Comparing neutralizing antibodies obtained from convalescent patients revealed that S2 targeting antibodies exhibit a lower neutralizing effect than those specific to the receptor binding domain in S1.
回復期患者から採取した中和抗体を比較すると、S2を標的とする抗体は、S1の受容体結合ドメインに特異的な抗体よりも中和効果が低いことが明らかになった。
5.2. Spike protein constructs in vaccine design
ワクチン設計におけるスパイクタンパク質構造
Current vaccines use different strategies to induce a robust immune response against the spike protein.
現在のワクチンは、スパイクタンパク質に対する強力な免疫反応を誘発するために、さまざまな戦略を採用しています。
The two most prevalent mRNA vaccines (‘BNT162b2’ by Pfizer–BioNTech and ‘mRNA-1273’ by Moderna) encode for a full-length spike protein stabilized in the prefusion state by two distinct mutations (K986P and V987P), whilst another vaccine (‘CoronaVac’ by Sinovac) employs the strategy of a wild-type spike protein present on the viral surface of the inactivated virus vaccine.
最も普及している 2 つの mRNA ワクチン (ファイザー・ビオンテックの「BNT162b2」とモデルナの「mRNA-1273」) は、2 つの異なる変異 (K986P と V987P) によって融合前の状態で安定化された全長スパイクタンパク質をコードしていますが、もう 1 つのワクチン (シノバックの「CoronaVac」) は、不活化ウイルスワクチンのウイルス表面に存在する野生型スパイクタンパク質の戦略を採用しています。
Novavax has recently completed a phase 3 trial showing strong efficacy for a vaccine (NVX-CoV2373) that only contains spikes as the antigen.
Novavax 社は最近、抗原としてスパイクのみを含むワクチン (NVX-CoV2373) の強力な有効性を示すフェーズ 3 試験を完了しました。
The contained spikes were produced with the same two proline changes present in the mRNA sequences, but also contain three changes to the ‘furin’ cleavage site rendering it unrecognizable.
含まれているスパイクは、mRNA 配列に存在するのと同じ 2 つのプロリン変化で生成されましたが、認識できないようにする「フーリン」切断部位の 3 つの変化も含まれています。
Structures in the prefusion state with the RBD either in ‘up’ or ‘down’ conformation are a focus of vaccine development, due to immunogenicity of the RBD epitopes.
RBD が「上」または「下」のコンフォメーションにある融合前状態の構造は、RBD エピトープの免疫原性のため、ワクチン開発の焦点となっています。
Many of the constructs used for vaccine design contain two mutated amino acids at positions 986 and 987, which help to stabilize the spike in the prefusion state.
ワクチン設計に使用される構造の多くは、位置 986 と 987 に 2 つの変異アミノ酸を含んでおり、融合前状態でスパイクを安定化するのに役立ちます。
However, these are now believed to alter the protein structure relative to the wild-type, as through the artificial mutation K986P a salt bridge between Lys986 and either Asp427 or Asp428 of an adjacent protomer is broken, leading to a relaxed apex.
ただし、現在では、人工変異 K986P によって Lys986 と隣接するプロトマーの Asp427 または Asp428 の間の塩橋が壊れ、弛緩した頂点につながるため、これらは野生型と比較してタンパク質構造を変化させると考えられています。
In a recent publication, the authors raised concerns that the altered structure could have an impact on vaccine efficiency, as antibodies induced by the spike with a relaxed apex may not recognize spikes presented on a viral surface.
最近の論文で、著者らは、弛緩した頂点を持つスパイクによって誘導される抗体はウイルス表面に存在するスパイクを認識しない可能性があるため、構造の変化がワクチンの効率に影響を与える可能性があるという懸念を表明した。
Hence, for vaccine design, structures of proteins in prefusion state without such kind of stabilizing mutations would presumably be preferred.
したがって、ワクチン設計では、このような安定化変異のない融合前状態のタンパク質の構造がおそらく好ましいでしょう。
Nevertheless, the currently used vaccines, Pfizer-BioNTech's ‘BNT162b2’ and Moderna's ‘mRNA-1273’, both using a stabilized prefusion spike, show a > 90% efficacy against wild-type SARS-CoV-2 after the second vaccination and six month follow up.
それにもかかわらず、現在使用されているワクチン、ファイザー-ビオンテックの「BNT162b2」とモデルナの「mRNA-1273」はどちらも安定化融合前スパイクを使用しており、2回目のワクチン接種と6か月間の追跡調査の後、野生型SARS-CoV-2に対して90%を超える有効性を示しています。
Several studies highlight the contributions of structural biology in the ongoing efforts to combat COVID-19, addressing challenges related to vaccine design and emerging variants.
いくつかの研究では、COVID-19と闘うための継続的な取り組みにおいて、ワクチン設計や新たな変異体に関連する課題に対処する構造生物学の貢献が強調されています。
5.3. Mutations and immune evasion
突然変異と免疫回避
Since the first occurrence of SARS-CoV-2 in Wuhan in 2019, the virus genome has repeatedly changed due to genetic drift.
2019年に武漢でSARS-CoV-2が初めて発生して以来、ウイルスのゲノムは遺伝的浮動により繰り返し変化してきました。
A genomic analysis of 10,333 spike protein sequences taken from samples from across the globe revealed that there are 400 distinct mutation sites in the spike protein, and 44 in the receptor binding domain alone.
世界中のサンプルから採取した 10,333 個のスパイクタンパク質配列のゲノム解析により、スパイクタンパク質には 400 個の異なる変異部位があり、受容体結合ドメインだけでも 44 個の変異部位があることが明らかになりました。
Another site of frequent mutation is the region around the protease cleavage site (between residues 675-692).
頻繁に変異が起こるもう 1 つの部位は、プロテアーゼ切断部位の周辺領域 (残基 675-692 の間) です。
Structural data of the full-length prefusion ectodomain is also used to facilitate evaluation of mutations in different viral strains including notable variants of concern such as the Delta variant (B.1.1617.2) and the Omicron Variant (B.1.1.529).
全長の融合前エクトドメインの構造データは、Delta 変異体 (B.1.1617.2) や Omicron 変異体 (B.1.1.529) などの注目すべき変異体を含むさまざまなウイルス株の変異の評価を容易にするためにも使用されます。
Depending on the location, each mutation impacts the spike conformational dynamics, receptor binding affinity or antigenicity, or combinations thereof.
場所に応じて、各変異はスパイクの立体構造ダイナミクス、受容体結合親和性または抗原性、またはそれらの組み合わせに影響を及ぼします。
For Omicron and Delta, recent studies suggest that due to their mutations, both strains are less sensitive to neutralizing monoclonal antibodies from recovered patients and capable of evading immunity conferred through prior infections.
オミクロンとデルタについては、最近の研究で、その変異により、回復した患者から得られる中和モノクローナル抗体に対する感受性が低く、以前の感染によって付与された免疫を回避する能力があることが示唆されています。
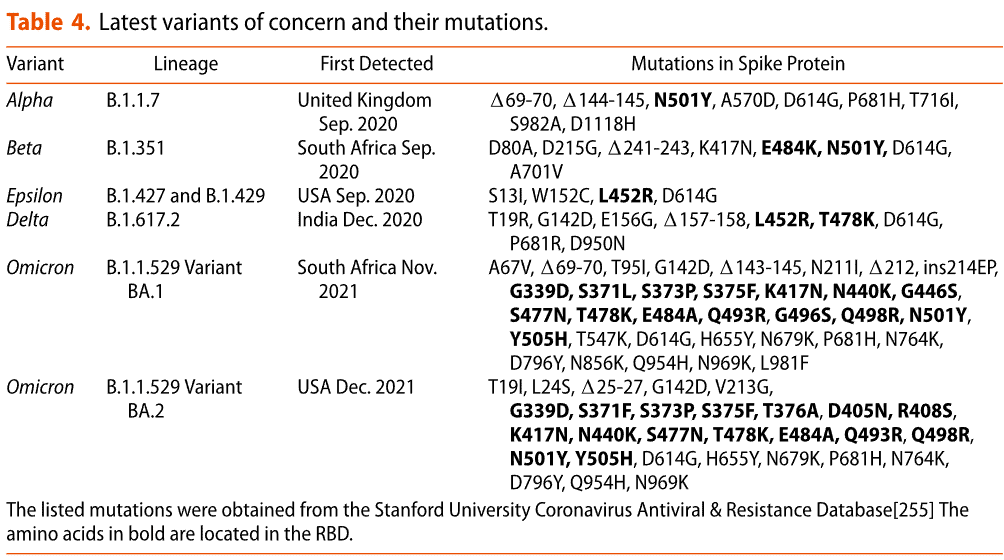
An overview of some of the latest variants of concern and interest and their mutations in the spike protein is given in Table 4.
懸念と関心の対象となっている最新の変異体と、それらのスパイクタンパク質の変異の概要を表 4 に示します。
PDB codes related to latest virus variants are summarized in Table 5.
最新のウイルス変異体に関連する PDB コードは表 5 にまとめられています。
The spike protein of the Delta variant contains eight mutations and two deletions and the variant is reported to be twice as infectious as the wild-type.
デルタ変異体のスパイクタンパク質には 8 つの変異と 2 つの欠失が含まれており、この変異体は野生型の 2 倍の感染力があると報告されています。
Mutation T19R and the deletion 156 and 157 are located in the N-termina domain ‘supersite’ which is the target of most anti-NTD antibodies.
変異 T19R と欠失 156 および 157 は、ほとんどの抗 NTD 抗体の標的である N 末端ドメイン「スーパーサイト」にあります。
Mutation L452R and T478K, located in the receptor binding domain, have been identified as potentially significant for the infectivity and transmissibility of the virus.
受容体結合ドメインにある変異 L452R および T478K は、ウイルスの感染性と伝染性に潜在的に重要であることが確認されています。
A simulation study noted a difference in the receptor-binding interface between the wild type and a L452R/T478K variant: A receptor-binding β-loop-β motif adopts an altered conformation, leading to shifts in epitope regions and a reduction in antibody recognition.
シミュレーション研究では、野生型と L452R/T478K 変異体との間の受容体結合インターフェースに違いがあることが指摘されました:受容体結合 β-ループ-β モチーフは変化した構造をとり、エピトープ領域の変化と抗体認識の低下につながります。
In contrast to the Delta variant, the Omicron variant is much more heavily mutated and includes up to 30 mutations in the spike protein relative to the wild-type (Wuhan-Hu-1), depending on the precise sub-variant (BA.1 or BA.2).
デルタ変異体とは対照的に、オミクロン変異体は変異がはるかに激しく、野生型(武漢-Hu-1)と比較してスパイクタンパク質に最大30の変異が含まれており、正確なサブ変異体(BA.1またはBA.2)によって異なります。
Of those mutations, approximately half are located in the RBD and 11 are involved in receptor binding.
これらの変異のうち、約半分は RBD にあり、11 は受容体結合に関与しています。
These mutations substantially affect the binding mode to ACE2 and its interaction with antibodies and drugs developed against the wild-type spike protein.
これらの変異は、ACE2への結合モードと、野生型スパイクタンパク質に対して開発された抗体および薬剤との相互作用に大きく影響します。
To date, it seems that Omicron is twice as infectious as Delta and more than ten times as infectious as the wild type.
これまでのところ、オミクロンはデルタの2倍、野生型の10倍以上の感染力があるようです。
Key residues proposed to contribute in a critical way to the enhanced affinity of Omicron to ACE2 include mutations T478K and N501Y, which were also present in prior variants (T487K: Delta; N501Y: Alpha, Beta) and N440K.
オミクロンのACE2に対する親和性の向上に決定的な形で寄与すると提案されている主要な残基には、以前の変異体(T487K:デルタ、N501Y:アルファ、ベータ)にも存在していた変異T478KとN501Y、およびN440Kが含まれます。
Mutations K417N, E484A and Y505H are involved in disrupting binding of antibodies to the spike, leading to an indicated 14-fold higher vaccine escape capability than Delta.
K417N、E484A、Y505H の変異は、スパイクへの抗体の結合を阻害する働きがあり、デルタよりも 14 倍高いワクチン回避能力が示されています。
If infection rates remain high, new variants of SARS-CoV-2 with further mutations, most likely in the RBD or NTD, will emerge quickly.
感染率が高いままであれば、RBD または NTD にさらなる変異が生じた SARS-CoV-2 の新しい変異体が急速に出現するでしょう。
Antibodies which compete with RBD-ACE2 binding are under some of the strongest selection pressure from variants.
RBD-ACE2 結合と競合する抗体は、変異体から最も強い選択圧を受けています。
Over the course of the pandemic, mutations have developed which favour either improved binding with the ACE2 receptor (often through electrostatic complementarity), or escape from existing antibody responses and many which do both (such as L452R, seen in the lineages B.1.427, B.1.429 (Epsilon), Delta and then again in the Omicron-variant (Table 4)).
パンデミックの過程で、ACE2受容体との結合の改善(多くの場合、静電的相補性による)または既存の抗体反応からの回避のいずれかに有利な変異が開発され、その両方を行う変異も多くありました (B.1.427、B.1.429(イプシロン)、デルタ、そして再びオミクロン変異体(表4)に見られるL452Rなど)。
Occasionally, mutations seem to balance improved infectivity with an increased susceptibility to the human immune system, such as K417N.
時には、K417Nのように、変異によって感染性の向上とヒト免疫システムに対する感受性の増加がバランスしているようです。
Dramatically different sets of antibodies are raised against different variants.
異なる変異体に対しては、劇的に異なる抗体セットが生成されます。
The original wild-type SARS-CoV-2 strain elicited a strong V3-53 (variable gene) sequence response which was not seen against the Beta variant (Table 4), which moderately favoured antibodies with V4-39 sequence.
元の野生型 SARS-CoV-2 株は、V3-53 (可変遺伝子) 配列に対する強い反応を引き起こしましたが、ベータ変異体 (表 4) に対しては見られませんでした。ベータ変異体では、V4-39 配列を持つ抗体がやや優勢でした。
ACE2-competing antibodies raised against these variants, which only differed by K417N, E484K and N501Y in the RBD, were found to be completely distinguishable in cluster analysis using neutralization data.
これらの変異体に対して生成された ACE2 競合抗体(RBD の K417N、E484K、および N501Y のみが異なる)は、中和データを使用したクラスター分析で完全に区別できることが判明した。
This demonstrates how sensitive the antibody response is to mutations in this region.
これは、この領域の変異に対する抗体応答の感度の高さを示している。
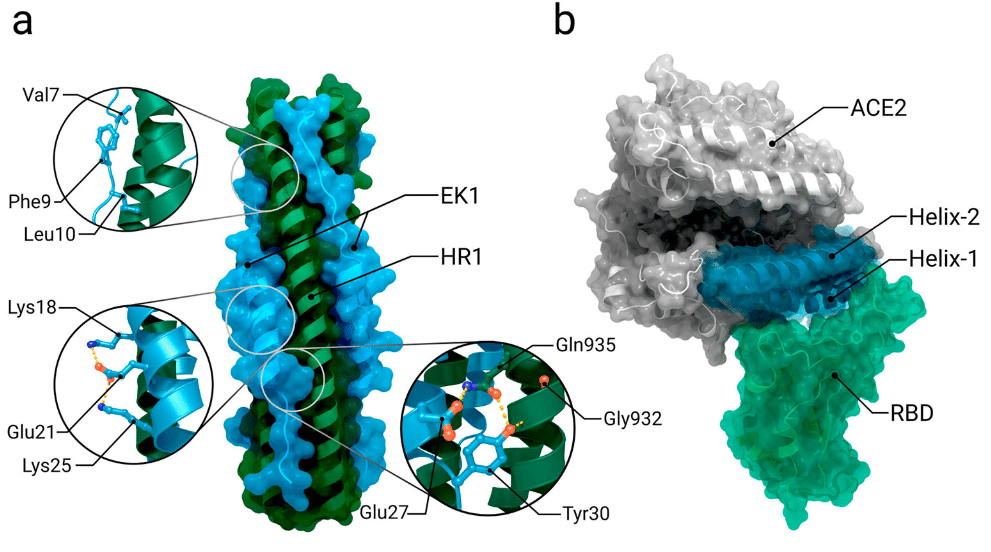
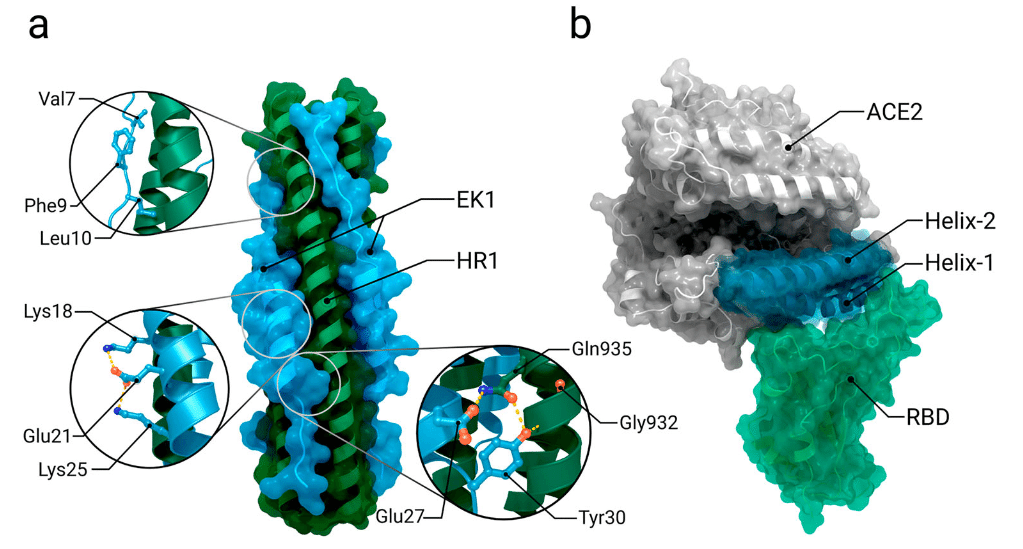
A significant opportunity for spike binding inhibition is through the use of soluble human ACE2 derivates, which bind to the viral RBDs and thereby prevent any interaction with target cell receptors (Figure 7b).
スパイク結合阻害の大きな可能性は、可溶性ヒトACE2誘導体の使用です。これはウイルスRBDに結合し、それによって標的細胞受容体との相互作用を防止します(図7b)。
An advantage of this approach is its intrinsic resilience to viral mutation escape: though some viral mutations may result in a reduced antibody recognition, selection leading to a higher affinity towards ACE2 will increase the potency of these ‘ACE2 traps’.
このアプローチの利点は、ウイルス変異の回避に対する本質的な耐性です:一部のウイルス変異は抗体認識の低下につながる可能性がありますが、ACE2に対する親和性を高める選択により、これらの「ACE2トラップ」の効力が高まります。
As interaction with the spike mainly occurs at the N-terminal α-helices of ACE2, potential ACE2 derivates include those helices; such mimetics have already been shown to be successful in neutralizing SARS-CoV-1.
スパイクとの相互作用は主に ACE2 の N 末端 α ヘリックスで起こるため、潜在的な ACE2 誘導体にはこれらのヘリックスが含まれます。このような模倣物は SARS-CoV-1 の中和に効果があることがすでに示されています。
It has also been suggested that inclusion of the N-terminal domain in addition to the receptor binding domain as a target for neutralizing antibodies in vaccine development could broaden the number of neutralizing epitopes and reduce the potential of the virus to escape host immunity.
また、ワクチン開発において、受容体結合ドメインに加えてN末端ドメインを中和抗体の標的として含めることで、中和エピトープの数が増え、ウイルスが宿主免疫から逃れる可能性が減る可能性もあると示唆されている。
Conversely, it seems that the NTD has been subjected to high evolutionary pressure as almost all variants of concern have extensive mutations around these sites.
逆に、NTD は、懸念されるほぼすべての変異体がこれらの部位の周囲に広範な変異を持っていることから、高い進化圧力にさらされているようです。
Analysis of sequence data from COG-UK (Covid-19 Genomics UK Consortium) shows that the NTD has an even higher mutation rate and that no clear consensus for specific mutations amongst notable variants has emerged, though there is a tendency towards reduction of the overall NTD mass.
COG-UK(Covid-19 Genomics UK Consortium)の配列データの分析では、NTDの変異率がさらに高く、注目すべき変異体の間で特定の変異に関する明確なコンセンサスは出ていないものの、NTD全体の質量は減少傾向にあることが示されています。
This feature would putatively make the NTD a poor target for escape-resistant therapeutics.
この特徴により、NTDは逃避耐性治療薬のターゲットとしては適さないと考えられます。
5.4. Molecular inhibitors
分子阻害剤
In addition to neutralizing antibodies, numerous molecular inhibitors are currently under development to block cell entry via the spike protein, encompassing fusion inhibitors and protease inhibitors.
中和抗体に加えて、融合阻害剤やプロテアーゼ阻害剤を含む、スパイクタンパク質を介した細胞侵入を阻止するための多数の分子阻害剤が現在開発中です。
It's important to note that this review provides only a very limited selection of examples, while several publications offer a more comprehensive overview of different molecular inhibitors.
このレビューではごく限られた例しか提供されていないことに注意することが重要ですが、いくつかの出版物ではさまざまな分子阻害剤のより包括的な概要が提供されています。
Given that the spike protein facilitates virus-cell fusion, fusion inhibitors present a promising approach for preventing SARS-CoV-2 cell entry.
スパイクタンパク質がウイルスと細胞の融合を促進することを考えると、融合阻害剤は SARS-CoV-2 の細胞侵入を防ぐ有望なアプローチとなります。
To bring the viral and host cell membranes in close proximity for membrane fusion, spikes need to rearrange into a pre-hairpin intermediate.
ウイルスと宿主細胞の膜を膜融合のために近づけるには、スパイクをプレヘアピン中間体に再配置する必要があります。
During these structural rearrangements a six-helix bundle is formed between the trimeric coiled coil of HR1 and helical HR2 segments.
これらの構造再配置中に、HR1 の三量体コイルドコイルとらせん状の HR2 セグメントの間に 6 ヘリックス束が形成されます。
One approach to inhibiting the fusion process is for peptides sharing sequence identity to the heptad repeat 1 and 2 domains to bind the pre-hairpin intermediate, thereby preventing formation of the six-helix bundle and subsequent membrane fusion (Figure 7a).
融合プロセスを阻害する 1 つのアプローチは、ヘプタッド リピート 1 および 2 ドメインと配列同一性を共有するペプチドがプレヘアピン中間体に結合し、6 ヘリックス バンドルの形成とそれに続く膜融合を防ぐことです (図 7a)。
Another major strategy for targeting the spike protein involves protease inhibitors.
スパイクタンパク質を標的とするもう一つの主要な戦略は、プロテアーゼ阻害剤を使用することです。
The entry of SARS-CoV-2 into airway epithelial cells likely depends on proteolytic cleavage in the S1/S2 interface and within the S2′ site.
SARS-CoV-2 が気道上皮細胞に侵入するには、S1/S2 界面および S2′ 部位内でのタンパク質分解による切断が関係していると考えられます。
Priming of the S2′ site is supposed to be performed by type II transmembrane serine protease (TMPRSS2).
S2′ 部位のプライミングは、II 型膜貫通セリンプロテアーゼ (TMPRSS2) によって行われると考えられています。
Different TMPRSS2 inhibitors, such as camostat mesylate, nafamostat mesylate, and peptidomimetics, are among those being investigated for the treatment of COVID-19.
COVID-19の治療薬として研究されているものには、カモスタットメシル酸塩、ナファモスタットメシル酸塩、ペプチド模倣薬などのさまざまなTMPRSS2阻害剤があります。
Camostat mesylate and nafamostat mesylate are synthetic serine protease inhibitors targeting TMPRSS2 and related proteases.
カモスタットメシル酸塩とナファモスタットメシル酸塩は、TMPRSS2と関連プロテアーゼを標的とする合成セリンプロテアーゼ阻害剤です。
Camostat mesylate has been shown to significantly reduce but not completely abolish infection of Calu3 lung cells in vitro.
カモスタットメシル酸塩は、in vitroでCalu3肺細胞の感染を大幅に軽減しますが、完全には除去しないことが示されています。
The approval of camostat mesylate for human use in Japan highlights its potential as a promising drug.
カモスタットメシル酸塩が日本でヒトへの使用が承認されたことは、有望な薬剤としての可能性を浮き彫りにしています。
Important to note is that in cell culture, camostat mesylate is rapidly converted into GBPA (4-(4-guanidinobenzoyloxy) phenylacetic acid), which also has TMPRSS2 inhibitory activity, albeit to a lesser extent.
注目すべき重要な点は、細胞培養において、カモスタットメシル酸塩は急速に GBPA (4-(4-グアニジノベンゾイルオキシ)フェニル酢酸) に変換され、これも TMPRSS2 阻害活性を持つが、その程度は低いことである。
Potential and highly promising drug alternatives to camostat mesylate are peptidomimetics.
カモスタットメシル酸塩の潜在的かつ非常に有望な代替薬はペプチド模倣薬である。
N-0385, a peptidomimetic with inhibitory activity against TMPRSS2 has shown to effectively block SARS-CoV-2 infection in Calu-3 cells.
TMPRSS2に対する阻害活性を持つペプチド模倣薬であるN-0385は、Calu-3細胞におけるSARS-CoV-2感染を効果的に阻止することが示されています。
When administered to K18-hACE2 mice infected with SARS-CoV-2, N-0385 effectively prevented mortality caused by the virus and significantly mitigated pulmonary damage.
SARS-CoV-2に感染したK18-hACE2マウスに投与したところ、N-0385はウイルスによる死亡を効果的に予防し、肺損傷を大幅に軽減しました。
In the same study, the authors state that N-0385 could be a promising drug for the early treatment of disease caused by the SARS-CoV-2 wild type and delta variant.
同研究で、著者らは、N-0385 が SARS-CoV-2 野生型およびデルタ変異体によって引き起こされる疾患の早期治療に有望な薬剤となる可能性があると述べています。
The inhibitors mentioned here represent just a subset of the promising TMPRSS2 inhibitors available, with numerous others extensively documented in a comprehensive literature review by Wettenstein et al.
ここで言及した阻害剤は、現在入手可能な有望な TMPRSS2 阻害剤のほんの一部に過ぎず、Wettenstein らによる包括的な文献レビューでは、他にも多数の阻害剤が詳細に文書化されています。
6. Summary
The SARS-CoV-2 spike plays a crucial role during pathogenesis of COVID-19, as it mediates entry of SARS-CoV-2 into host cells.
SARS-CoV-2 スパイクは、SARS-CoV-2 の宿主細胞への侵入を媒介するため、COVID-19 の発症に重要な役割を果たします。
It consists of two subunits, S1 and S2, each of which contains several domains needed for interaction with the host cell and subsequent membrane fusion.
これは S1 と S2 の 2 つのサブユニットで構成され、それぞれに宿主細胞との相互作用とそれに続く膜融合に必要ないくつかのドメインが含まれています。
The hinge-like movement of the receptor binding domain from the ‘down’ to the ‘up’ conformation is crucial for receptor binding, since this movement exposes the interaction epitope.
受容体結合ドメインが「下」から「上」の構造にヒンジのように動くことは、相互作用エピトープを露出させるため、受容体結合に重要です。
However, this epitope is also a main target for neutralizing antibodies.
ただし、このエピトープは中和抗体の主なターゲットでもあります。
To evade the immune system and reduce interactions with antibodies, the RBD of the spike can switch into the down conformation.
免疫系を回避し、抗体との相互作用を減らすために、スパイクの RBD は下の構造に切り替えることができます。
Cleavage with type II transmembrane serine protease (TMPRSS2) after receptor binding induces shedding of subunit S1 and triggers S2 to undergo major structural rearrangements that project the fusion peptide towards the host cell membrane, resulting in formation of a pre-hairpin intermediate that bridges viral and host cell membrane.
受容体結合後のII型膜貫通セリンプロテアーゼ(TMPRSS2)による切断により、サブユニットS1の脱落が誘発され、S2は主要な構造再配置を受け、融合ペプチドを宿主細胞膜に向けて突き出し、ウイルスと宿主細胞膜を橋渡しするプレヘアピン中間体の形成を引き起こします。
The membranes are then brought into close proximity for fusion through formation of a six-helix bundle by the heptad repeat 1 and 2 domains.
その後、ヘプタッドリピート1および2ドメインによる6ヘリックス束の形成により、膜は融合のために近接します。
A striking feature of the spike is its glycan shield, which protects about 60% of its surface from being recognized by antibodies.
スパイクの顕著な特徴は、そのグリカンシールドで、表面の約60%が抗体によって認識されるのを防ぎます。
Nevertheless, all currently used vaccines are based on the spike, which is the most attractive vaccine target, mainly due to its exposed location.
それにもかかわらず、現在使用されているすべてのワクチンはスパイクに基づいています。スパイクは、主に露出している場所のために、最も魅力的なワクチンターゲットです。
7. Discussion & outlook
Since 2019, over 800 protein structures related to the SARS-CoV-2 spike protein have been deposited in the PDB.
2019年以降、SARS-CoV-2スパイクタンパク質に関連する800以上のタンパク質構造がPDBに登録されています。
This is emblematic of the amount of collective research that has been conducted worldwide on the coronavirus and its proteins during the pandemic.
これは、パンデミック中に世界中でコロナウイルスとそのタンパク質について行われた共同研究の量を象徴しています。
The contributions of structural biology and biophysics to the fight against COVID19 have been compiled and presented in a number of works.
COVID19との戦いにおける構造生物学と生物物理学の貢献は、多くの研究でまとめられ、発表されています。
The spike protein is one of the most studied proteins of SARS-CoV-2 and the collection of available structures provides a comprehensive overview of its function and antigenicity.
スパイクタンパク質は、SARS-CoV-2 で最も研究されているタンパク質の 1 つであり、利用可能な構造のコレクションにより、その機能と抗原性の包括的な概要が提供されます。
They also serve as a fundamental basis for drug and vaccine development.
これらは、薬剤およびワクチン開発の基本的な基盤としても機能します。
Nevertheless, the spike is a large complex and many questions remain open regarding intra- and intermolecular interactions with different proteins, such as different proteases, cell membranes and the immune system.
とはいえ、スパイクは大きな複合体であり、さまざまなプロテアーゼ、細胞膜、免疫システムなどのさまざまなタンパク質との分子内および分子間相互作用に関して多くの疑問が残っています。
To date there is no full-length structure of the spike inserted into the membrane (with cytoplasmic tail and transmembrane domain) and every ectodomain structure contains unresolved sections.
今のところ、膜に挿入されたスパイクの全長構造(細胞質尾部と膜貫通ドメインを含む)は存在せず、すべてのエクトドメイン構造には未解決のセクションが含まれています。
These presumably highly flexible/dynamic regions most commonly include the furin cleavage site, the heptad repeat 1 domain and especially the fusion peptide region.
これらのおそらく高度に柔軟/動的である領域には、通常、フーリン切断部位、ヘプタッドリピート1ドメイン、特に融合ペプチド領域が含まれます。
The absence of the latter limits the mechanistic understanding of intramolecular interactions as well as the understanding of the fusion process.
後者の不存在は、分子内相互作用のメカニズムの理解だけでなく、融合プロセスの理解も制限をもたらします。
In addition to coronaviridae, virus families that pose a danger to society such as orthomyxoviridae (influenza) and retroviridae (HIV) also utilise type-I-fusion proteins.
コロナウイルス科に加えて、オルトミクソウイルス科(インフルエンザ)やレトロウイルス科(HIV)など、社会に危険をもたらすウイルス科もI型融合タンパク質を利用しています。
Thus, the knowledge obtained through coronavirus research may be useful in gleaning further insights into the fusion mechanism of other viruses and thereby help to develop therapeutics and vaccines to prevent or limit future infections.
したがって、コロナウイルス研究を通じて得られた知識は、他のウイルスの融合メカニズムに関するさらなる洞察を得るのに役立ち、将来の感染を予防または制限するための治療薬やワクチンの開発に役立つ可能性があります。
Since the first appearance of SARS-CoV-2, its genome has changed many times through mutations, resulting in multiple new variants of the virus.
SARS-CoV-2 が初めて出現して以来、そのゲノムは突然変異によって何度も変化し、その結果、ウイルスの複数の新しい変異体が生まれました。
One of the proteins most affected by mutations is the spike protein, due to the combination of the high replication rate and high selection pressure from immunization by infection or vaccines.
突然変異の影響を最も受けやすいタンパク質の 1 つは、高い複製率と感染またはワクチンによる免疫化による高い選択圧の組み合わせによるスパイクタンパク質です。
Under these circumstances, mutations emerge that balance affinity and binding of the human cell surface receptor ACE2 with evading the immune system.
このような状況下では、ヒト細胞表面受容体 ACE2 の親和性と結合と免疫システムの回避とのバランスをとる突然変異が出現します。
An example of this is the Omicron variant, whose heavily mutated spike protein results in the highest transmissibility and most effective immune evasion, where it is not uncommon for individuals to be reinfected within relatively short time periods.
その一例がオミクロン変異体で、スパイクタンパク質が大きく突然変異すると、最も高い伝染性と最も効果的な免疫回避がもたらされ、比較的短期間で再感染することも珍しくありません。
Given the importance of the up–down–up-down dynamics of the receptor binding domain for evading the immune system, there remains significant scope for characterising the structural effects of different mutations on these dynamics, which may reveal mechanisms for modifying them.
免疫システムを回避するための受容体結合ドメインの上下上下ダイナミクスの重要性を考えると、これらのダイナミクスに対するさまざまな変異の構造的影響を特徴付ける大きな余地が残っており、それによって、それらを変更するメカニズムが明らかになる可能性があります。
What makes this difficult is that mutations may not ultimately have a significant impact on the average position of atoms within a model, but rather the relative stability of different (sub)states or interactions; identification of these features requires us to go beyond the analysis of average atomic positions.
これを困難にしているのは、変異が最終的にはモデル内の原子の平均位置に大きな影響を与えるのではなく、さまざまな(サブ)状態または相互作用の相対的な安定性に影響を与える可能性があることです;これらの特徴を特定するには、平均原子位置の分析を超える必要があります。
As the coronavirus SARS-CoV-2 will become endemic, mutations in the spike protein will likely have a long-term impact on societies as they continue to change SARS-CoV-2’s pathogenicity.
コロナウイルス SARS-CoV-2 がエンデミック化するにつれ、スパイクタンパク質の変異は SARS-CoV-2 の病原性を変化させ続け、社会に長期的な影響を及ぼす可能性があります。
Continuous study of the spike domains of new variants is therefore essential for pandemic preparedness, i.e. to adjust the composition of vaccines so that effective antibodies are created that efficiently interfere with membrane fusion of new SARS-CoV-2 variants and human airway epithelial cells.
したがって、新しい変異体のスパイクドメインの継続的な研究は、パンデミックへの備え、つまり、新しい SARS-CoV-2 変異体とヒトの気道上皮細胞の膜融合を効率的に阻害する効果的な抗体が生成されるようにワクチンの組成を調整することに不可欠です。
As long as there are reservoirs where SARS-CoV-2 can amplify, the spike protein will collect new mutations in antibody-binding sites, leading to changes in antibody recognition.
SARS-CoV-2 が増幅できるリザーバーがある限り、スパイクタンパク質は抗体結合部位に新しい変異を集め、抗体認識の変化につながります。
Hence, it is the properties of the spike that may well determine the future course and the ultimate outcome of the pandemic.
したがって、パンデミックの今後の進路と最終的な結果を決定づけるのは、スパイクの特性である可能性があります。
It is therefore vital that we keep up with structural changes in new variants as they emerge.
したがって、新しい変異体が出現するたびに、その構造変化に遅れずについていくことが重要です。
以下省略。
この記事が気に入ったらサポートをしてみませんか?