
【研究】RNA-seq04:発現比較解析準備編(RとRStudioの導入)

こんにちは、あるいは、こんばんは。
相変わらずの山積みの雑務をこなしつつ、ようやく発現比較解析に手を付けています。
マッピング後のカウント処理(【研究】RNA-seq03)は、実行できていないため記述できておりません。
カウントデータファイルがありますので、統計ソフト「R」を統合開発環境(IDE)の「RStudio」を用いて解析しています。
PCスペック
MacBook Pro 14-inch, 2021
チップ:Apple M1 Max
コア:10(パフォーマンス 8, 効率性: 2)
メモリ:64 GB
macOS:macOS Sonoma 14.3
RとRStudio
生命科学の分野に携わると、いつか手を出す時期がくるのが「R」と思います。もちろん、「Python」にも触ることがあると思います。
今回は「RStudio」を用いて、RNA-seqのカウントデータの解析を進めてみたいと思います。
Rのインストール
公式サイトからインストーラーをダウンロードし、インストールします。
バージョンによって、ライブラリが使用できない場合もあるため、解析が実行できる環境が構築できた場合には、バージョンを控えておくと良いようです。
RStudioのインストール
Rの統合開発環境(IDE)を利用すると、Rスクリプトの管理、データの読み書き、解析経過で作成されたデータの確認などが簡便になります。
公式サイトからインストーラーをダウンロードし、インストールします。
これで、Rでの解析準備ができましたね。
Rの実行
「R」と「RStudio」をインストールしたら、「RStudio」を起動します。
基本的な使い方は、「R入門」などで検索すると学習できるサイトがたくさんヒットすると思います。
最近では、「ChatGPT」を教師として、質問を投げかけると教えてくれると思いますよ。
Rの練習
ChatGPTに教えていただきます。
Rの基本的な使い方について、
解析に使用できるようなRNA-seqによる遺伝子発現データ(遺伝子シンボル、各群のカウント値、遺伝子のアノテーション情報などを含める)を定義して、
1)基本的な情報の表示
2)データの操作と抽出
3)データの集計
4)データの統計解析
5)データの可視化
6)データの解釈
について例題を示して教えていただけますか。
理解しました。以下は、RNA-seqによる遺伝子発現データのサンプルを作成し、それを使って基本的な使い方を例示するコードです。
# 遺伝子発現データのサンプル作成
gene_symbols <- c("Gene1", "Gene2", "Gene3", "Gene4", "Gene5")
group1_counts <- c(100, 150, 80, 200, 120)
group2_counts <- c(90, 160, 70, 180, 110)
group3_counts <- c(110, 140, 90, 190, 130)
annotation_info <- c("Function1", "Function2", "Function3", "Function4", "Function5")
# データフレームの作成
gene_expression_data <- data.frame(GeneSymbol = gene_symbols,
Group1_Counts = group1_counts,
Group2_Counts = group2_counts,
Group3_Counts = group3_counts,
Annotation = annotation_info)ここまでを左上ペイン「Untitled1*(Rスクリプトのこと)」にコピペして、実行したいコード部分を選択し、「Run」をクリック(または「⌘ + ↩️」)して実行します。
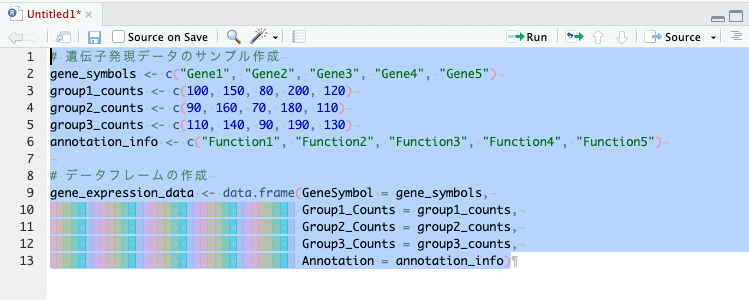
左下のペイン「コンソール」にコードが入力され、実行されます。
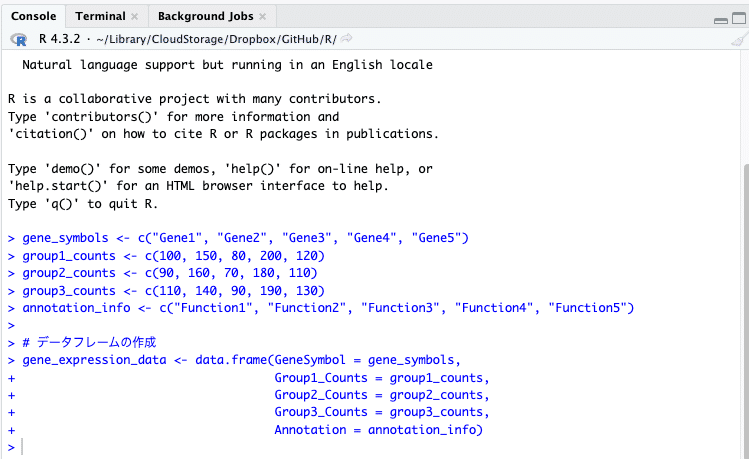
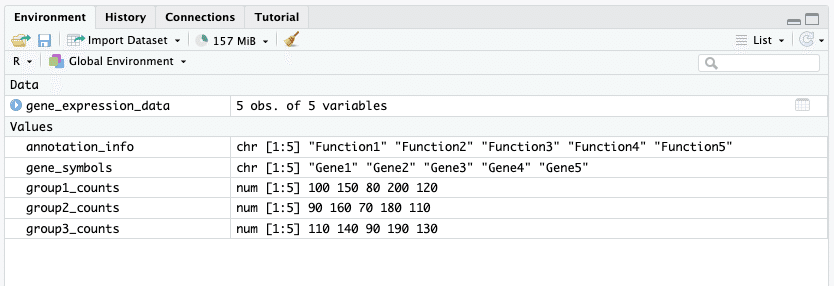
右上ペイン「Environment」に、「Data」と「Values」が追加され、先ほどのコードで定義した情報が記録されます。
同様に、以下のコードを左上ペインにコピーして、実行します。
# 1)基本的な情報の表示
print("基本的な情報の表示:")
print(summary(gene_expression_data))
print(head(gene_expression_data))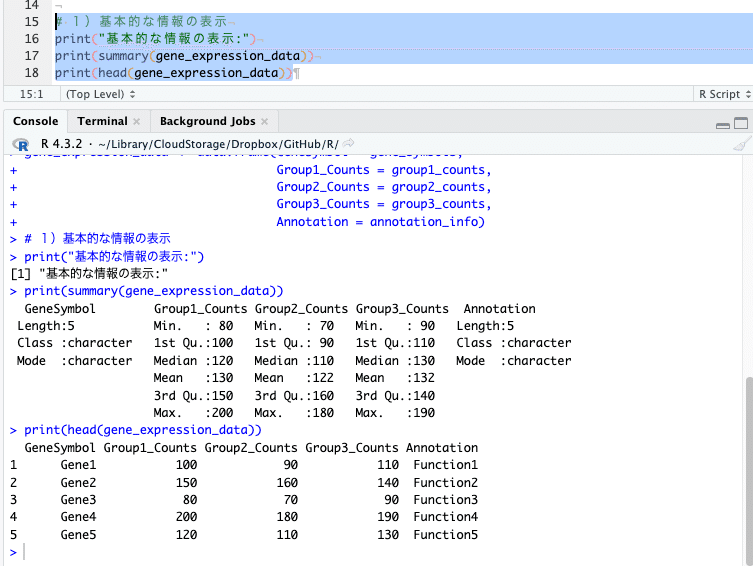
左下ペイン「コンソール」に、基本的な情報として、統計サマリーと上から6行分のデータ(今回は5行分しかデータがない)を表示してくれます。
続けて、以下のコードを左上ペインにコピーして、実行します。
# 2)データの操作と抽出
print("\nデータの操作と抽出:")
# Gene1のデータを抽出
print("Gene1のデータ:")
print(gene_expression_data[gene_expression_data$GeneSymbol == "Gene1", ])
# グループ1のカウント値が150以上の遺伝子を抽出
print("グループ1のカウント値が150以上の遺伝子:")
print(gene_expression_data[gene_expression_data$Group1_Counts >= 150, ])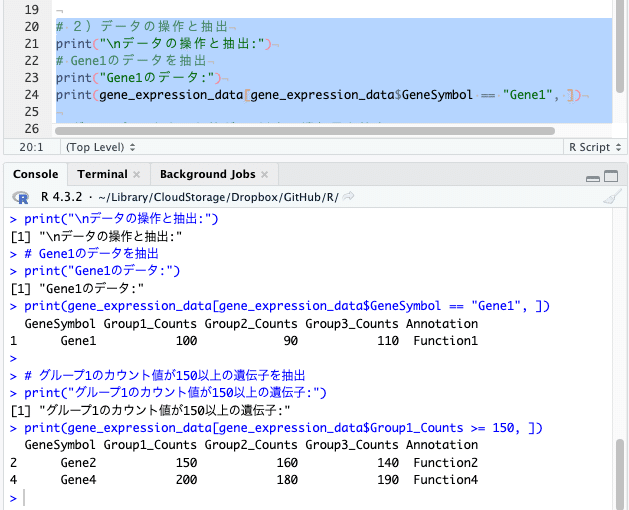
様々な条件のデータ抽出を行うことができます。
以下、同様に以下のコードを左上ペインにコピーして、実行していきます。
# 3)データの集計
print("\nデータの集計:")
# 各グループの平均カウント値を計算
average_counts <- colMeans(gene_expression_data[, 2:4])
print("各グループの平均カウント値:")
print(average_counts)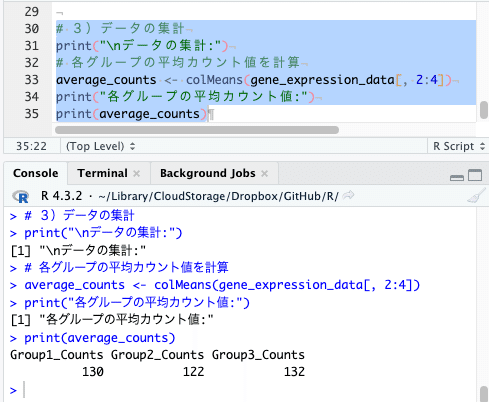
各グループの平均カウント値を計算して、「average_counts」に格納していますね。
続けます。
# 4)データの統計解析
print("\nデータの統計解析:")
# グループ間のt検定を行う
t_test_result <- t.test(gene_expression_data$Group1_Counts, gene_expression_data$Group2_Counts)
print("グループ1とグループ2のt検定結果:")
print(t_test_result)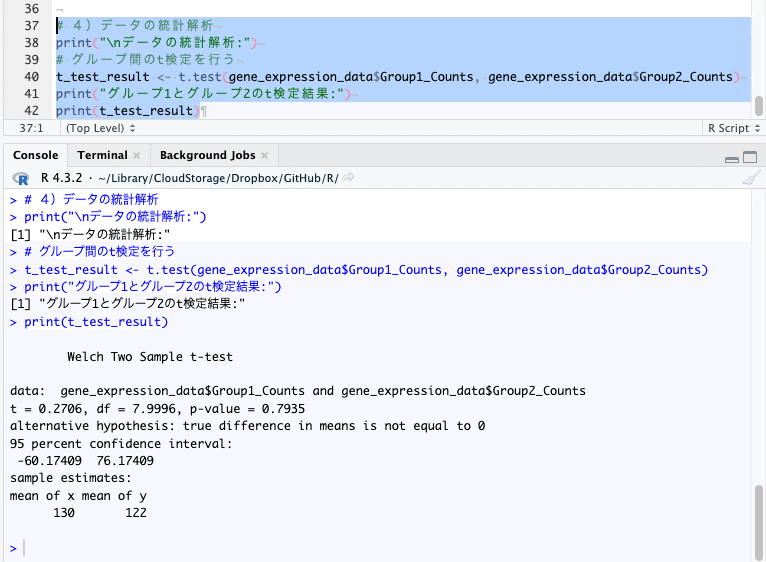
グループ1とグループ2のt検定を行っています。
p-valueは0.7935 ( > 0.05 ) です。
グラフを描画します。
# 5)データの可視化
print("\nデータの可視化:")
# グループ間の発現量の比較をボックスプロットで表示
boxplot(gene_expression_data[, 2:4], col = c("red", "blue", "green"), main = "Gene Expression Comparison")
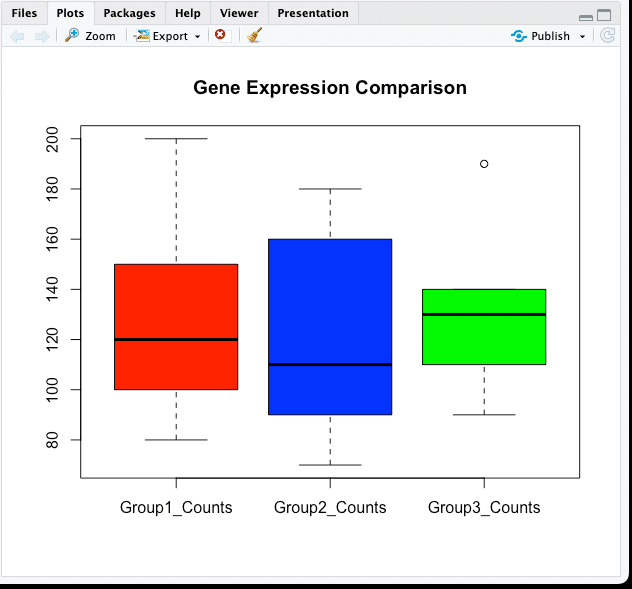
左下ペイン「コンソール」には何も結果は表示されず、右下「Plots」ペインにグラフが表示されます。
最後にデータの解釈です。これは「R」の機能というより、「ChatGPT」によるデータの解釈(?)を表示させるコードです。
# 6)データの解釈
print("\nデータの解釈:")
print("グループ1とグループ2の間に有意な遺伝子発現量の差が見られることが示唆されました。")
「4)データの統計解析」で確認したように(p > 0.05)、「グループ1とグループ2の間に有意な遺伝子発現量の差が見られることが示唆されました。」というのは誤った情報となります。
まとめ
いかがでしょうか。
コードの作成をリクエストすると、ある程度、処理可能な内容で提案してくれます。
望み通りのコードになっていなくても、少し修正すると使える内容になることもありますので、困ったときに相談してみると良いかもしれません。
ただし、嘘(誤り)の情報をあたかも真(正解)の情報として返してきたり、作動しないコードや複雑難解なコードを返してきたりもします。
Google先生にも聞いて、自分自身で情報を整理・取捨選択することも重要です。

更新
20240228:初版
この記事が気に入ったらサポートをしてみませんか?