
【研究】RNA-seq02:マッピング編v20240229.0

こんにちは、あるいは、こんばんは。
執筆作業、会議資料作成、会議もろもろ、ようやく一時のオフィス籠もりタイムができましたので、格闘した格闘中の成果をご報告します。
(2024/2/9追記, 2/29追記)
PCスペック
MacBook Pro 14-inch, 2021
チップ:Apple M1 Max
コア:10(パフォーマンス 8, 効率性: 2)
メモリ:64 GB
macOS:macOS Sonoma 14.3
Apple silicon M1でなんとか動かそうと奮闘しておりましたが、
結論、「無理!」
でした。使用できないツールが多く、なんとか動かそうと努力しましたが、対応していないので無理なようです。
Rosetta 2でターミナル.appを動かしております。
今回の目標
前回までに、解析に使用するデータの確認まで済ませました。
● 【研究】RNA-seq01:準備編 : 今回、環境リセットします
● 【研究】RNA-seq01.5:納品データ確認編 : データ使用します
今回は実データを用いて解析の前処理をし始めます!
解析環境については、今後、使用する可能性がありますが、基本的にリセットして一から再構築します。
ちなみに。。。
Intel CPUのMacBook Proを持っておりますが、そちらでも動くと思います。
(途中まで動作を確認しております)
MacBook Pro 13-inch, 2016, Four Thunderbolt 3 Ports
Processor:3.3 GHz Dual-Core Intel Core i7
Memory:16 GB 2133 MHz LPDDR3
Graphics:Intel Iris Graphics 550 1536 MB
macOS:macOS Monterey 12.7.3
環境構築
今までに構築した環境とは異なる環境を使用していきます。
もし、これまでにいろいろなプログラムをご使用の場合には、バックアップを取るなどしてから実施してください。
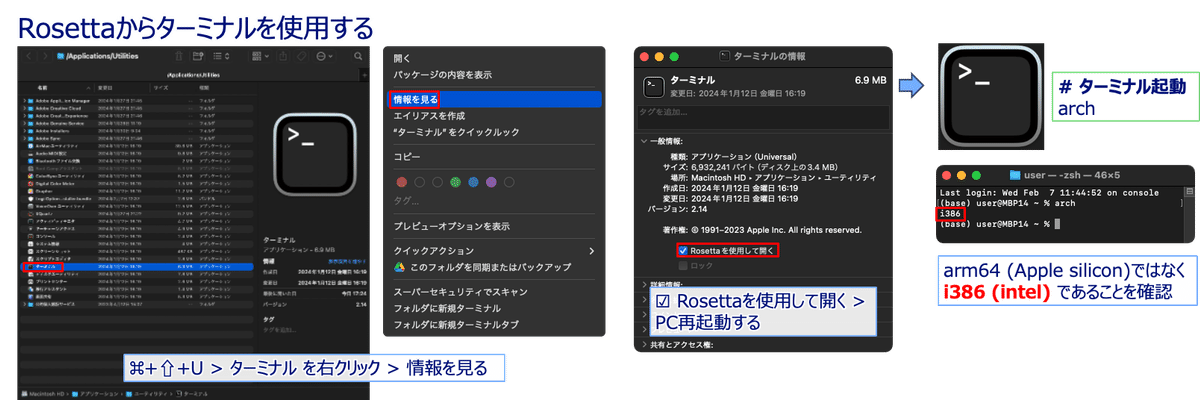
Rosetta 2でターミナル.appを使用する
・「⌘+⇧+U」(/Applications/ユーティリティ)
・「ターミナル」を右クリック
・ 情報を見る
・ Rosettaを使用して開く
・ PCを再起動する
・ ターミナルを起動する
・ 以下のコマンド「arch」を実行
arch
# > i386i386と返ってくれば、Intel CPUを使用していることと同じ挙動でターミナルを使用できます。
もし、Rosettaなんて見つからない!という場合には
これまで、Rossetaを使用する機会がなかった場合には、チェックボックスがないかもしれません(詳しくはわからないです)
そのような時には、以下のコードを実行してみてください。
(おまじないです)
softwareupdate --install-rosettaCondaをインストール
今回は、「Mambaforge3」というものを導入してみます。
Rosetta2から起動したターミナルに以下のコードを入力します。
ワーキングディレクトリは、好きな場所で良いと思います。
今回、dropboxではなく、外付けSSDまたはSDXCを使用しています。
少しずつ変更がありますが、ご容赦ください。
# ワーキングディレクトリの変更: ここにダウンロードされます
cd /Volumes/1TB/RNAseq/03
# GitHubから「Mambaforge3.sh」としてダウンロードします
wget -O Mambaforge3.sh https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-MacOSX-x86_64.sh
# インストールします
bash Mambaforge3.sh -b
# パスを通します
echo ". ~/mambaforge/etc/profile.d/conda.sh" >> ~/.bashrc && \
echo "conda activate base" >> ~/.bashrc
source ~/.bashrcもし、「wget」がないなどのエラーがありましたら、以前の記事を参考にしてHomebrewを導入していただければと思います。
以下の記事の中に記載があります。
Chnannel設定
Biocondaが推奨するchannel設定をしておきます。
以下のコードを順番に実行します。
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
conda config --set channel_priority strictツールのインストール
解析の前処理などに使用するツールをインストールします。
コンフリクトも起こるようで、わたしには解決ができません。
もし、不具合が発生するようでしたら、環境をリセットし、その時に必要なツールのみインストールすると良いと思います。
mamba install -c bioconda seqkit -y # 追記2/29
mamba install -c bioconda bowtie2 -y
mamba install -c bioconda sra-tools -y
mamba install -c bioconda samtools -y
mamba install -c bioconda fastp -y
mamba install -c bioconda fastqc -y
mamba install -c bioconda fastq-screen -y
mamba install -c bioconda hisat2 -y
mamba install -c bioconda stringtie -y
mamba install -c conda-forge pigz -y
mamba install libtiff pillow simplejson matplotlib -y # <- multiqcで必要
mamba install -c bioconda multiqc -y # 実施しました2/9
mamba install -c bioconda igv -y # 起動確認2/9
mamba install -c bioconda igvtools -y # 起動確認2/9multiqcを実施してみました。関連パッケージのインストールが必要になるとのことでした。(2024/2/9追記)
seqkitを実施してみました。(2024/2/29追記)
データの配置
データの配置は皆様それぞれと思いますが、コードを実行する前に、ワーキングディレクトリを確認すると良いと思います。
何も考えず、シンプルに動かす場合には、全てワーキングディレクトリに保存してしまいましょう。
また、必要なデータやファイルがワーキングディレクトリと別に保存されている場合には、その都度、指定するようにお願いします。
データ保存場所のイメージ

FASTAの簡易解析(2024/2/29追記)
seqkitを用いて、FASTAの簡易的な解析を行います。
以下のコードを実行します。
seqkit stats /Volumes/1TB/RNAseq/rawdata/*.fastq.gzオプションも様々あり、SeqKitでは色々な操作ができるようです。詳細はSeqKitの公式ページをご確認ください。
-a、--all:すべての統計情報を出力
-b、--basename:塩基名のみを出力
-E、--fq-encoding string:fastqの品質エンコーディングを指定
-G、--gap-letters string:ギャップを表す文字を指定
-h、--help:統計情報に関するヘルプを表示
-e、--skip-err:エラーをスキップし、警告メッセージのみ表示
-S、--skip-file-check:指定ファイルのチェックをスキップ
-T、--tabular:タブ形式で出力 など
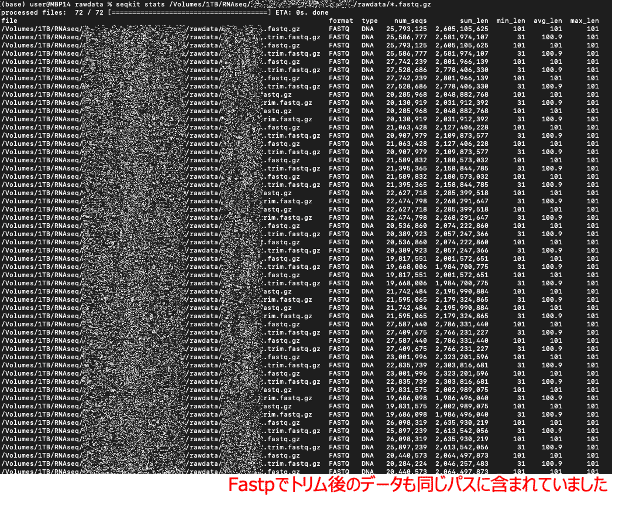
以下が表示されます。72データの解析に約2時間30分かかりました。
Fastpでトリムした後のデータも同じパスに含まれていたため、時間がかかってしまいました。
念の為、コピーしてテキストデータとして保存しておきます。
トリミング
Fastp
Fastpを用いてアダプター配列などのトリミングを行います。
深く理解して処理を進めているわけではないため、別にTrimmomaticでの処理も検討いたします(2024/2/29追記)。
まず、Fastpで処理を行います。
以下のコードを実行します。
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/rawdata
# サンプルリストファイルのパス
SAMPLE_LIST_FILE="sample_list.txt"
## サンプルリストを読み込んで処理するループ
## NR > 1: 1行目(header)を無視する
awk 'NR > 1 {print $2}' "$SAMPLE_LIST_FILE" | while IFS= read -r SAMPLE; do
echo "Processing sample: $SAMPLE"
# fastpの実行
fastp --detect_adapter_for_pe \
-i "${SAMPLE}_1.fastq.gz" \
-I "${SAMPLE}_2.fastq.gz" \
-o "${SAMPLE}_1.trim.fastq.gz" \
-O "${SAMPLE}_2.trim.fastq.gz" \
-h "${SAMPLE}_report.html" -w 1
echo "Sample $SAMPLE processing complete."
done詳細はChatGPTに確認する必要がありますが、ループを利用してサンプルリストに記載のあるIDとシーケンスデータ名と照合して、トリミングを実行します。
サンプルリストの1行目には項目名があり、その部分を飛ばして処理します(NR > 1)。2列目にはサンプルIDがあり、データ名と照合する情報です(awk '{print $2}')
fastpのオプションは以下の通りです。
-i:リード1の入力データ
-I:リード2の入力データ
-o:リード1の出力データ
-O:リード2の出力データ
-h:出力レポート
-w:スレッド数
以下のデータ・レポートが出力されます。
# トリムされたデータ
……_1.trim.fastq.gz
……_2.trim.fastq.gz
# レポートファイル
……_report.html
fastp report (.html) の例

Trimmomatic(2024/2/29追記)
Trimmomaticも実行してみました。
本法ではアダプター配列を指定して処理を進めることになります。
以下のコードを実行します。
# アダプター配列情報のダウンロード
cd /Users/user/Downloads
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
unzip Trimmomatic-0.39.zip
# Trimmomaticのインストール
mamba install -c bioconda trimmomatic -y
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/rawdata
# サンプルリストファイルのパス
SAMPLE_LIST_FILE="sample_list.txt"
## サンプルリストを読み込んで処理するループ
## NR > 1: 1行目(header)を無視する
awk 'NR > 1 {print $2}' "$SAMPLE_LIST_FILE" | while IFS= read -r SAMPLE; do
echo "Processing sample: $SAMPLE"
# trimmomaticの実行
trimmomatic PE -threads 10 -trimlog ${SAMPLE}.txt \
${SAMPLE}_1.fastq.gz ${SAMPLE}_2.fastq.gz \
paired_${SAMPLE}_1.trim.fastq.gz unpaired_${SAMPLE}_1.trim.fastq.gz \
paired_${SAMPLE}_2.trim.fastq.gz unpaired_${SAMPLE}_2.trim.fastq.gz \
ILLUMINACLIP:/Users/user/Downloads/Trimmomatic-0.39/adapters/TruSeq2-PE.fa:2:30:10
echo "Sample $SAMPLE processing complete."
done試しに1サンプルのみ処理してみましたが、約1時間40分かかりました。
以下のデータが出力されます。
# ログファイル
sample1.txt
# トリムされたデータ
paired_sample1_1.trim.fastq.gz
paired_sample1_2.trim.fastq.gz
unpaired_sample1_1.trim.fastq.gz
unpaired_sample1_2.trim.fastq.gz
この処理をループですべて実行するには、、、、約30時間(18データ)かかることになります意外にも約5時間で終わりました!
後述のマッピング処理では、コードの内容に「paired_」を加えて処理すれば良さそうですね。
(2024/2/29追記ここまで)
クオリティチェック(QC)
FastQC
FastQCを用いてデータの質(クオリティ)の確認をします。
以下のコードを実行します。
fastqc --nogroup -o . *.trim.fastq.gz以下のデータ・レポートが出力されます。
# データ
……_1.trim_fastqc.zip
……_2.trim_fastqc.zip
# レポートファイル
……_1.trim_fastqc.html
……_2.trim_fastqc.html
fastqc report (.html) の例
この続きは実行中のため、後日追記いたします。
ようやく解析のスタートラインが見えてきました。
(2024/2/7 20:50)
MultiQC
MultitQCを用いてデータの質(クオリティ)の確認をします。
「/Volumes/1TB/RNAseq/rawdata」に格納されている「fastp report」と「fastqc report」を認識して、まとめて提示してくれます。
以下のコードを実行します。
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/rawdata
# MultiQC実行
multiqc .以下のフォルダ・レポートが出力されます。
# フォルダ
multiqc_data
- multiqc.log
- multiqc_data.json
- multiqc_fastp.txt
- multiqc_sources.txt
- multiqc_general_stats.txt
- multiqc_citations.txt
- multiqc_software_versions.txt
- multiqc_fastqc.txt
# レポートファイル
multiqc_report.html
multiqc report (.html) の例
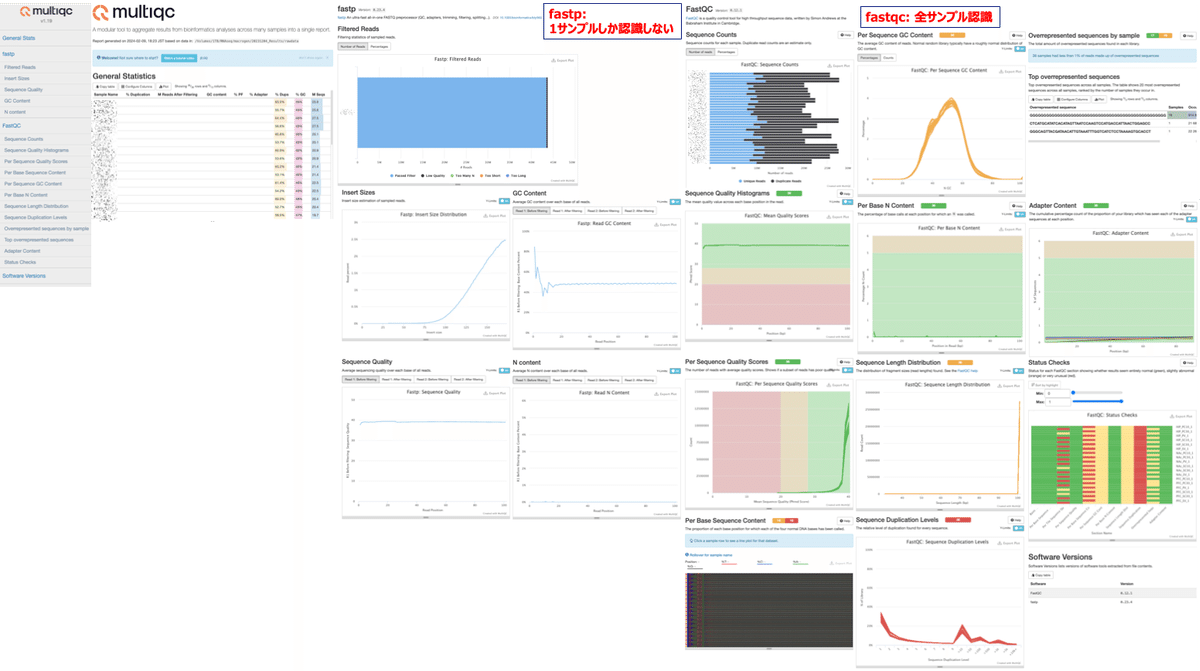
fastqcのレポートは全36レポートを認識してくれた
マッピング
HISAT2
HISAT2を用いてリードデータのマッピングします。
まずリファレンスゲノム配列のインデックスを準備しておきます。
作成するディレクトリは、リードデータと同じ場所でも大丈夫です。
今回は「cd /Volumes/1TB/RNAseq/03」に格納しています。
以下のコードを実行します(約40分)。
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/03
# リファレンスゲノム配列のダウンロード
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/annotation_releases/10090/109/GCF_000001635.27_GRCm39/GCF_000001635.27_GRCm39_genomic.fna.gz
# 解凍
unpigz -d GCF_000001635.27_GRCm39_genomic.fna.gz
# index作成(約40分)
hisat2-build GCF_000001635.27_GRCm39_genomic.fna index_GRCm39_genomic以下の8つのインデックスが出力されます。
# インデックス
index_GRCm39_genomic.1.ht2
index_GRCm39_genomic.2.ht2
index_GRCm39_genomic.3.ht2
index_GRCm39_genomic.4.ht2
index_GRCm39_genomic.5.ht2
index_GRCm39_genomic.6.ht2
index_GRCm39_genomic.7.ht2
index_GRCm39_genomic.8.ht2
作成したインデックスを使用して、リードデータをマッピングします。
以下のコードを実行します(時間がかかるためO/N:19:00〜)。
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/rawdata
# サンプルリストファイルのパス
SAMPLE_LIST_FILE="sample_list.txt"
## サンプルリストを読み込んで処理するループ
## NR > 1: 1行目(header)を無視する
awk 'NR > 1 {print $2}' "$SAMPLE_LIST_FILE" | while IFS= read -r SAMPLE; do
echo "Processing sample: $SAMPLE"
# hisat2 (mapping)の実行
FILE1=${SAMPLE}_1.trim.fastq.gz
FILE2=${SAMPLE}_2.trim.fastq.gz
INDEX=/Volumes/1TB/RNAseq/03/index_GRCm39_genomic
hisat2 -p 1 -x ${INDEX} -1 ${FILE1} -2 ${FILE2} \
| samtools sort -@ 1 > ${SAMPLE}.sorted.bam
done以下のデータ「.bam」が出力されます。
# データ
…….sorted.bam
bamファイルのインデックス
bamファイルの情報を検索するための索引となります。
以下のコードを実行します。
# ワーキングディレクトリ
cd /Volumes/1TB/RNAseq/rawdata
# サンプルリストファイルのパス
SAMPLE_LIST_FILE="sample_list.txt"
## サンプルリストを読み込んで処理するループ
## NR > 1: 1行目(header)を無視する
awk 'NR > 1 {print $2}' "$SAMPLE_LIST_FILE" | while IFS= read -r SAMPLE; do
echo "Processing sample: $SAMPLE"
# SAMtools (index)の実行
samtools index -@ 1 ${SAMPLE}.sorted.bam
done 以下のデータ「.bai」が出力されます。
# データ
…….sorted.bam.bai
BAMとBAI
BAM(Binary Alignment Map)とBAI(Binary Alignment Index)は、DNAやRNAの配列データを保存するファイル形式
通常、BAMとBAIはペアで使用し、アラインメントデータの効率的な操作と解析に使用する
⚫︎BAM
リード配列(DNA・RNA断片)のアラインメント情報のバイナリ
リード配列、クオリティスコア、アラインメント位置、アラインメント方向などの情報を含む
⚫︎BAI
BAMに含まれる情報を高速に検索するための索引(インデックス)
BAMファイル内のリード位置情報を含む
特定のリードやリード範囲をBAMファイルから効率的に取得できる
マッピング結果の確認
IGV(Integrative Genomics Viewer)
IGVを用いて、リードデータの可視化を行います。
以下のコードを実行します。
ivgIVGが起動しました。
試しにgenome情報をダウンロードしてみました。
Genomes > Select Hosted Genome… > 「Mouse mm39」


BAMとBAIを読み込ませると良いのだと思いますが、上手くいきませんでした。勉強不足です。いつの日か、更新したいと思います。
更新
20240207.0:初版
20240209.0:MultiQC, マッピング(HISAT2, SAMtools)追記
20240229.0:SeqKit, Trimmomatic追記
この記事が気に入ったらサポートをしてみませんか?