塩野義製薬が開発中のCOVID-19治療薬「S-217622」について、こんな少ないデータで申請するなんて信じられない!

塩野義製薬が2月7日、開発中の新型コロナウイルスの飲み薬について、来週末か再来週にも厚生労働省に承認申請する可能性があることを明らかにした。同社では去年9月から国内で軽症や無症状の患者およそ2000人を対象に最終段階の治験を実施しており、有効性や安全性について良好な結果が確認できれば、最終治験が完了する前に実用化を認める「条件付き早期承認制度」の適用を想定し、承認申請をする可能性があるという。
さて、この薬剤の有効性・安全性に関するデータが塩野義製薬からプレスリリースされており、その内容を紹介させて頂きながら、私見を述べさせて頂きます。
試験概要 / 早期第二相試験 (Phase 2a part)
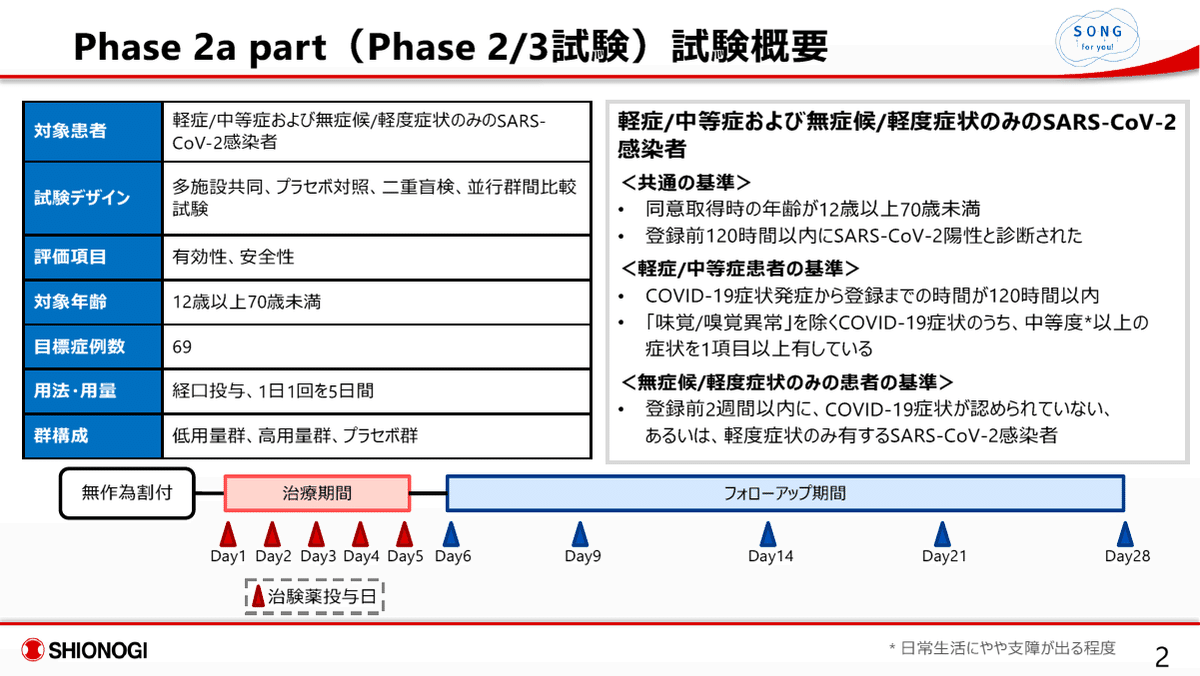
本試験は、S-217622の患者における有効性・安全性のあたりを観る試験であり、本格的に有効性・安全性を評価する前段階の臨床試験である。よって、この試験結果に基づいて、仮免許にせよ承認を与えることは従来はあり得ないことである。症例数も低用量群/16例、高用量群/14例、プラセボ群/17例と非常に少ない。

有効性の評価に関し、主要評価項目 (本試験における最も重要な有効性に関する評価指標) がウイルス力価の変化量となっており、これは臨床的な効果を確実に反映したものではない事。例えば、ウイルス力価が低下しても、低下した状態で症状が悪化するようなケースも考えられ、ウイルス力価と臨床症状との間の相関データが示されていない。後半に、PAXLOVID / Pfizer 及び Molnupiravir / Merck の承認取得に至った臨床試験における有効性の評価を示したが、これら2薬剤の有効性の評価は、患者の入院率や死亡率で評価されており、実際の臨床的有用性を反映した評価指標になっている。
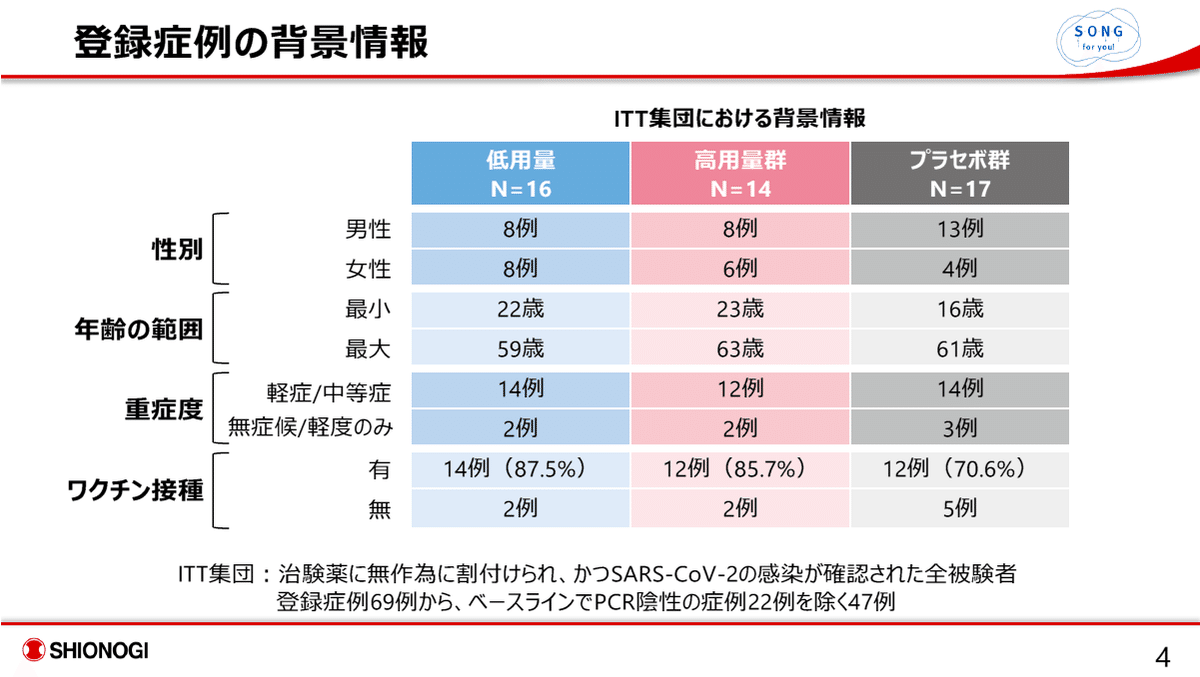
プラセボ群において、ワクチン無接種患者が5例と、薬剤投与群の各2例に比し、多い点が気になる。


プラセボ群においても、ウイルス力価、ウイルスRNA量は一貫して低下しており、有意差をもって薬剤投与群においてウイルス力価、ウイルスRNA量の低下は認められるものの、この結果が臨床的にどのような意義をもつのかが不明確である。

臨床的意義が無明確。

臨床的意義が不明確。

これが、唯一、臨床的な有効性を評価できるデータであるが、プラセボ群においても改善傾向が認められ、薬剤投与群において、より高い改善傾向は認められるが、プラセボ投与群との間に統計的有意差は認められていない。また、高用量群の方が低用量群よりウイルス力価の低下傾向は大きかったが、症状スコアにおいては、低用量群の方が改善傾向が大きい。単純に解釈すると、ウイルス力価と臨床症状との相関は低いのではないかとも考えられる。

ワクチン未接種例数がプラセボ群で5例、薬剤群では各2例であったので、その影響を無視できない。




薬剤投与群において、明らかに、副作用発現率が高い。しかも、用量依存性が垣間見える事から、安全性が高い薬剤だとは決して主張することはできない。


以上のように、塩野義製薬のS-217622を、上記データをもって、仮免許を与えるというのは、絶対に許されないと思われる。今、オミクロンの感染爆発により医療の逼迫状態が続いており、供給量をあまり心配する必要のない国産経口薬への期待の大きいのは理解できるが、本薬剤が仮に承認されれば、非常に多くの患者に投与されることになり、潜在的な投与患者数を考慮すると、たかだか60例程度の試験データで承認するようなことは、あってはならない事である。
PAXLOVID™ (nirmatrelvir and ritonavir) / Pfizer
Pfizerの臨床試験データの詳細 (最終結果)
NEW YORK--(BUSINESS WIRE) December 14, 2021 -- Pfizer Inc. (NYSE: PFE) today announced final results from an analysis of all 2,246 adults enrolled in its Phase 2/3 EPIC-HR (Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients) trial of its novel COVID-19 oral antiviral candidate PAXLOVID™ (nirmatrelvir and ritonavir). These results were consistent with the interim analysis announced in November 2021, showing PAXLOVID significantly reduced the risk of hospitalization or death for any cause by 89% compared to placebo in non-hospitalized, high-risk adult patients with COVID-19 treated within three days of symptom onset. In a secondary endpoint, PAXLOVID reduced the risk of hospitalization or death for any cause by 88% compared to placebo in patients treated within five days of symptom onset, an increase from the 85% observed in the interim analysis. The EPIC-HR data have been shared with the U.S. Food and Drug Administration (FDA) as part of an ongoing rolling submission for Emergency Use Authorization.
“This news provides further corroboration (実証するもの、裏付ける証拠/kərɑ̀bəréiʃən) that our oral antiviral candidate, if authorized or approved, could have a meaningful impact on the lives of many, as the data further support the efficacy of PAXLOVID in reducing hospitalization and death and show a substantial decrease in viral load. This underscores (強調する、明確に示す/ʌ̀ndərskɔ́r) the treatment candidate’s potential to save the lives of patients around the world,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “Emerging variants of concern, like Omicron, have exacerbated the need for accessible treatment options for those who contract the virus, and we are confident that, if authorized or approved, this potential treatment could be a critical tool to help quell the pandemic.”
EPIC-HR Final Results
In the final analysis of the primary endpoint from all patients enrolled in EPIC-HR, an 89% reduction in COVID-19-related hospitalization or death from any cause compared to placebo in <patients treated within three days of symptom onset> was observed, consistent with the interim analysis. In addition, a consistent safety profile was observed. 0.7% of patients who received PAXLOVID were hospitalized through Day 28 following randomization (5/697 hospitalized with no deaths), compared to 6.5% of patients who received placebo and were hospitalized or died (44/682 hospitalized with 9 subsequent deaths). The statistical significance (統計学的有意差) of these results was high (p<0.0001:有意差あり). In a secondary endpoint, PAXLOVID reduced the risk of hospitalization or death for any cause by 88% compared to placebo in <patients treated within five days of symptom onset>; 0.8% of patients who received PAXLOVID were hospitalized or died through Day 28 following randomization (8/1039 hospitalized with no deaths), compared to 6.3% of patients who received placebo (66/1046 hospitalized with 12 subsequent deaths), with high statistical significance (p<0.0001). Relative risk reduction was 94% in <patients 65 years of age or older, one of the populations at highest risk for hospitalization or death>; 1.1% of patients who received PAXLOVID were hospitalized through Day 28 (1/94 hospitalized with no deaths), compared to 16.3% of patients who received placebo (16/98 hospitalized with 6 deaths), with high statistical significance (p<0.0001). In the overall study population through Day 28, no deaths were reported in patients who received PAXLOVID as compared to 12 (1.2%) deaths in patients who received placebo.
In the EPIC-HR trial, in a secondary endpoint, SARS-CoV-2 viral load at baseline and Day 5 have been evaluated for 499 patients. After accounting for baseline viral load, geographic region, and serology status, PAXLOVID reduced viral load by approximately 10-fold, or 0.93 log10 copies/mL, relative to placebo, indicating robust activity against SARS-CoV-2 and representing the strongest viral load reduction reported to date for an oral COVID-19 agent.
Treatment-emergent adverse events were comparable between PAXLOVID (23%) and placebo (24%), most of which were mild in intensity. Fewer serious adverse events (1.6% vs. 6.6%) and discontinuation of study drug due to adverse events (2.1% vs. 4.2%) were observed in patients dosed with PAXLOVID, compared to placebo, respectively.
All other secondary endpoints for this study were not yet available for this review. Full study data are expected to be released later this month and submitted to a peer-reviewed publication.
Molnupiravir / Merck
Molnupiravir, [one of two antiviral pills (Merck / Molnupiravir, Pfizer / Paxlovid) that have caused excitement in the past few months because preliminary clinical-trial results showed that they can significantly reduce hospitalizations and deaths from COVID-19], has yet to receive an emergency use authorization from the US Food and Drug Administration (FDA). An FDA advisory committee met on 30 November, and narrowly voted to recommend the drug candidate’s emergency approval by 13 to 10.
The agency's lengthy deliberations could signal uncertainties about the antiviral’s efficacy and safety: full trial data submitted to the FDA suggest that molnupiravir is less effective than originally thought, dampening scientists’ hopes that the relatively cheap and easy-to-administer treatment might change the course of the pandemic.
The results, released ahead of the advisory committee meeting, showed that the antiviral, [which was developed by the pharmaceutical firm Merck, based in Kenilworth, New Jersey, and the biotechnology company Ridgeback Biotherapeutics in Miami, Florida], decreased the risk of hospitalization from COVID-19 by 30% — down from a 50% reduction observed early in the trial. “That’s not all that good,” says Katherine Seley-Radtke, [a medicinal chemist who develops antiviral drugs at the University of Maryland, Baltimore County]. “It’s pretty lacklustre (パッとしない/lǽklʌ̀stə).” Eliav Barr, [the senior vice president of global medical and scientific affairs at Merck], says that a reduction in hospitalizations would still be beneficial, especially in areas that are experiencing a surge in infections.
Monoclonal antibody treatments, by contrast, reduce the risk of severe COVID-19 by up to 85%. But they are costly and need to be administered intravenously, so finding an effective oral antiviral for the disease has been a high priority for scientists around the world who want to be better able to treat high-risk patients in rural areas and under-resourced countries, Seley-Radtke says.
Merck’s initial study group included 762 people who received 4 pills of either the antiviral or a placebo twice a day, for 5 consecutive days, between May and early August. A second group included 646 people who received the same treatment between August and early October. All of the participants, [nearly 80% of whom were located in Europe or Latin America], started the regimen within five days of experiencing COVID-19 symptoms. For each group, researchers tracked the participants and measured how many ended up in hospital or died because of COVID-19 complications. In the first group, participants’ rate of hospitalization or death dropped by half if they took molnupiravir rather than a placebo. But in the second group, there was almost no difference in outcome for those on the antiviral compared with those on the placebo.
この記事が気に入ったらサポートをしてみませんか?