
Ring (test 投稿)

□ ehtelescope;
Scientists have obtained the first image of a black hole, using Event Horizon Telescope observations of the center of the galaxy M87. The image shows a bright ring formed as light bends in the intense gravity around a black hole that is 6.5 billion times more massive than the Sun
□ FQXi:
"You cannot see a black hole but its shadow...We are looking at a region we have never seen before...We are looking at the gates of hell, the event horizon, the point of no return." #EHTBlackHole #Brussels Event Horizon Telescope collaboration
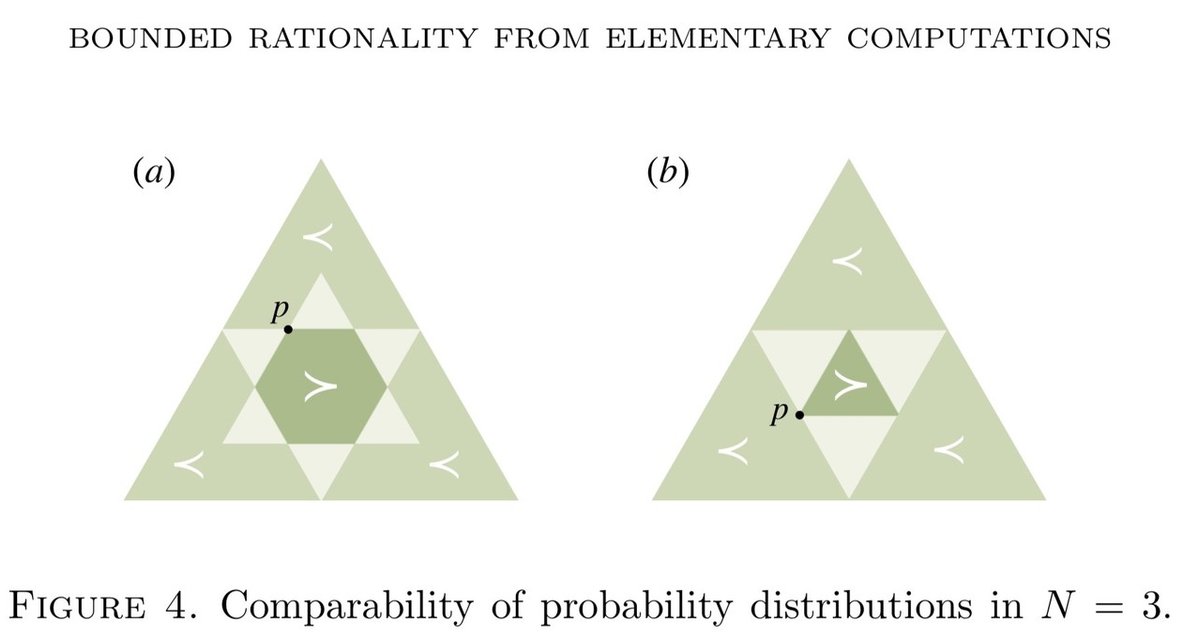
□ Bounded rational decision-making from elementary computations that reduce uncertainty
>> https://arxiv.org/pdf/1904.03964v1.pdf
Elementary computations can be considered as the inverse of Pigou- Dalton transfers applied to probability distributions, closely related to the concepts of majorization, T-transforms, and generalized entropies that induce a preorder on the space of probability distributions. As a consequence we can define resource cost functions that are order-preserving and therefore monotonic with respect to the uncertainty reduction.
This leads to a comprehensive notion of decision-making processes with limited resources. Along the way, they prove several new results on majorization theory, as well as on entropy and divergence measures.
□ Topological generation results for free unitary and orthogonal groups
>> https://arxiv.org/abs/1904.03974v1
every N≥3 the free unitary group U+N is topologically generated by its classical counterpart UN and the lower-rank U+N−1. This allows for a uniform inductive proof that a number of finiteness properties, known to hold for all N≠3, also hold at N=3. Specifically, all discrete quantum duals U+Nˆand O+Nˆare residually finite, and hence also have the Kirchberg factorization property and are hyperlinear.

□ Clairvoyante: A multi-task convolutional deep neural network for variant calling in single molecule sequencing
>> https://www.nature.com/articles/s41467-019-09025-z
Clairvoyante is the first method for Single Molecule Sequencing to finish a whole genome variant calling in two hours on a 28 CPU-core machine, with top-tier accuracy and sensitivity. Clairvoyante, a multi-task five-layer convolutional neural network model for predicting variant type, zygosity, alternative allele and Indel length.
□ Deep learning: new computational modelling techniques for genomics
>> https://www.nature.com/articles/s41576-019-0122-6
By effectively leveraging large data sets, deep learning has transformed fields such as computer vision and natural language processing. Now, it is becoming the method of choice for many genomics modelling tasks, including predicting the impact of genetic variation on gene regulatory mechanisms such as DNA accessibility and splicing.
□ Simulation of model overfit in variance explained with genetic data
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/10/598904.full.pdf
Pre-select SNPs on the basis of GWAS p<0.01 in the target sample. Enter target sample genotypes (the pre-selected SNPs) and phenotypes into an unsupervised machine learning algorithm (Phenotype-Genotype Many-to-Many Relations Analysis, PGMRA) for further reduction of the set of SNPs.
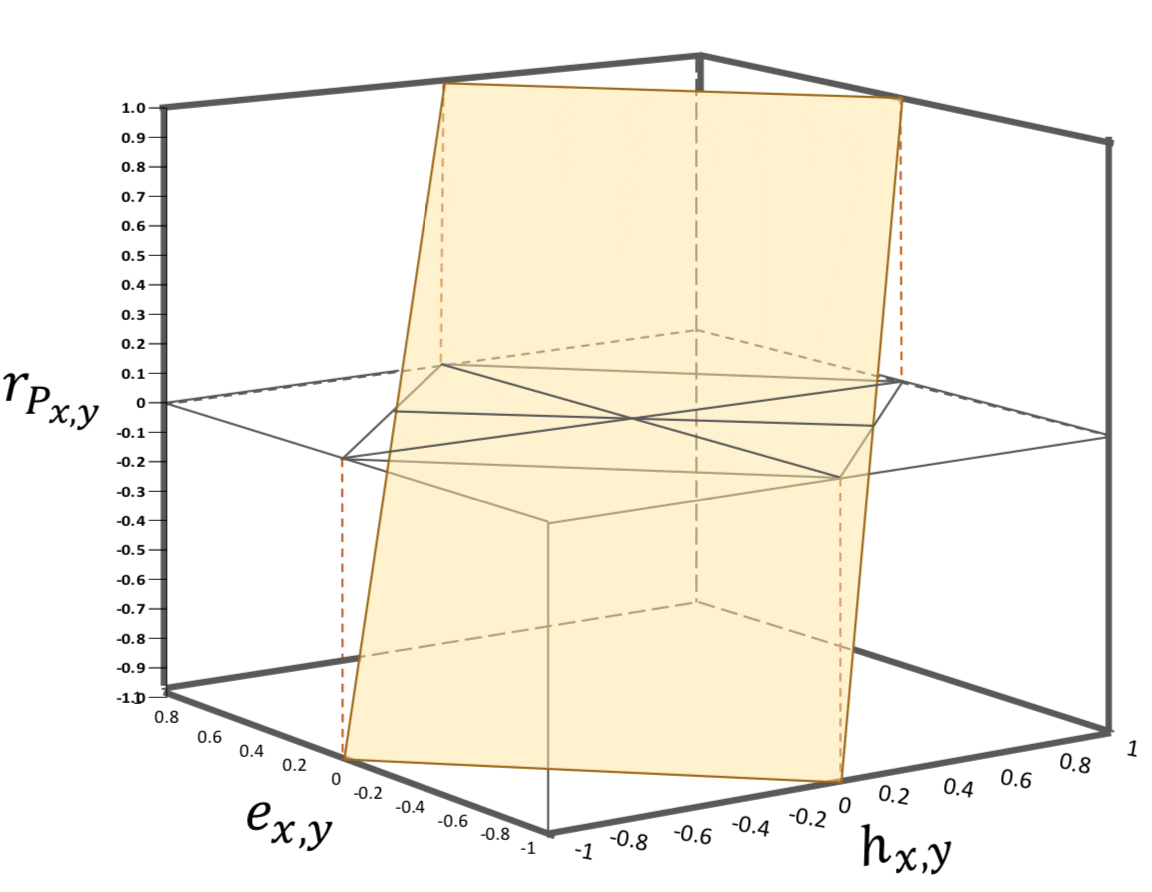
□ Coheritability and Coenvironmentability as Concepts for Partitioning the Phenotypic Correlation
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/10/598623.full.pdf
a mathematical and statistical framework is presented on the partition of the phenotypic correlation into these components. describing visualization tools to analyze the phenotypic correlation, coheritability and coenvironmentability concurrently, in the form of a three-dimensional (3DHER-plane) and a two-dimensional (2DHER-field) plots.
□ Malachite: A Gene Enrichment Meta-Analysis (GEM) Tool for ToppGene
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/10/511527.full.pdf
Malachite, a Python package that enables researchers to perform gene enrichment analyses on multiple gene lists and concatenate the resulting enrichment statistics. Malachite enables meta-enrichment analyses across multiple data sets.
To illustrate its use, we applied Malachite to three data sets from the Gene Expression Omnibus comparing gene expression. Biological processes enriched in all three data sets were related to xenobiotic stimulus.
□ Transport phenomena in bispherical coordinates
>> https://aip.scitation.org/doi/full/10.1063/1.5054581
This new bispherical equations are equally useful for setting up differential equations for new finite-difference solutions to transport problems.
the equations of change in bispherical coordinates cover a larger breadth of problems than previous work and allow for a unified approach to all future problems requiring exact solutions in bispherical or eccentric spherical systems.

□ The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight
>> https://science.sciencemag.org/content/364/6436/eaau8650.full
Presented here is an integrated longitudinal, multidimensional description of the effects of a 340-day mission onboard the International Space Station.
The persistence of the molecular changes (e.g., gene expression) and the extrapolation of the identified risk factors for longer missions (over 1 year) remain estimates and should be demonstrated with these measures in future astronauts.
□ Targeted Nanopore Sequencing with Cas9 for studies of methylation, structural variants and mutations
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/11/604173.full.pdf
the ability of this method to generate median 165X coverage at 10 genomic loci with a median length of 18kb from a single flow cell, which represents a several hundred fold improvement over the 2-3X coverage achieved without enrichment.
This technique has extensive clinical applications for assessing medically relevant genes and has the versatility to be a rapid and comprehensive diagnostic tool.
□ Stability index of linear random dynamical systems
>> https://arxiv.org/pdf/1904.05725v1.pdf
improving the Monte Carlo estimations by using certain linear constraints among the searched probabilities, take as final estimation of the searched probabilities the least squares solution of the inconsistent overdetermined system obtained when the Monte Carlo’s observed relative frequencies are forced to satisfy these linear constrains.
A suitable probability space, the starting point is to determine which is the “natural” election of the probability space and the distribution law of the coefficients of the linear dynamical system. Given a homogeneous linear discrete or continuous dynamical system, its stability index is given by the dimension of the stable manifold of the zero solution.
□ Boundary layer expansions for initial value problems with two complex time variables
>> https://arxiv.org/pdf/1904.04886v1.pdf
constructing inner and outer solutions of the problem and relate them to asymptotic representations via Gevrey asymptotic expansions with respect to ǫ, in adequate domains. The construction of such analytic solutions is closely related to the procedure of summation with respect to an analytic germ, whilst the asymptotic representation leans on the cohomological approach determined.
□ A learning-based framework for miRNA-disease association identification using neural networks
given a three-layer network, we apply a regression model to calculate the disease-gene and miRNA-gene association scores and generate feature vectors for disease and miRNA pairs based on these association scores.
given a pair of miRNA and disease, corresponding feature vector is passed through an auto-encoder-based model to obtain a low dimensional representation, and a deep convolutional neural network architecture is constructed.
□ A functional perspective on phenotypic heterogeneity in microorganisms
>> https://www.nature.com/articles/nrmicro3491
"Phenotypic heterogeneity is rather the rule than the exception"
□ The Michaelis-Menten paradox: Km is not an equilibrium constant but a steady-state constant.:
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/13/608232.full.pdf
The Michaelis-Menten constant (Km), the concentration of substrate ([S]) providing half of enzyme maximal activity, is higher than the ES → E S dissociation equilibrium constant. Actually, Km should be defined as the constant defining the steady state in the E S=ES → E P model and, accordingly, caution is needed when Km is used as a measure of the "affinity" of the enzyme-substrate interaction.
This paradox consists of the mechanistic meaning of Km in a dynamic framework. Km is equivalent in a dynamic situation to Kd in a static situation. Irrespective of the numeric values, K is the dissociation constant d (of the reaction E+S=ES) and Km is the steady-state constant.
□ fastGWA: A resource-efficient tool for mixed model association analysis of large-scale data
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/11/598110.full.pdf
fastGWA is an Mixed linear model (MLM)-based tool that controls for population stratification by principal components and relatedness by a sparse genetic relationship matrix for GWA analyses of biobank-scale data. fastGWA is robust in controlling for false positive associations in the presence of population stratification & relatedness, and that fastGWA is ~8x faster and only requires ~3% of RAM compared to the most efficient existing MLM-based GWAS tool in a very large sample (n=400,000).
□ Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF:
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/14/608869.full.pdf
a new iteration of Iterative Clustering and Guide-gene selection (ICGS) that outperforms state-of-the-art scRNA-Seq detection workflows when applied to well- established benchmarks.
This approach combines multiple complementary subtype detection methods (HOPACH, sparse-NMF, cluster “fitness”, SVM) to resolve rare and common cell- states, while minimizing differences due to donor or batch effects.
□ Sketching and Sublinear Data Structures in Genomics
>> https://www.annualreviews.org/doi/abs/10.1146/annurev-biodatasci-072018-021156
four key ideas that take different approaches to achieve sublinear space usage and processing time: compressed full text indices, approximate membership query data structures, locality-sensitive hashing, and minimizers schemes.

□ Comprehensive biodiversity analysis via ultra-deep patterned flow cell technology: a case study of eDNA metabarcoding seawater
>> https://www.nature.com/articles/s41598-019-42455-9
the NovaSeq detected many more taxa than the MiSeq thanks to its much greater sequencing depth. the pattern was true even in depth-for-depth comparisons. In other words, the NovaSeq can detect more DNA sequence diversity within samples than the MiSeq, even at the exact same sequencing depth.
These results are most likely associated to the advances incorporated in the NovaSeq, especially a patterned flow cell, which prevents similar sequences that are neighbours on the flow cell from being erroneously merged into single spots by the sequencing instrument.
□ Improving the sensitivity of long read overlap detection using grouped short k-mer matches
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5475-x
While using k-mer hits for detecting reads’ overlaps has been adopted by several existing programs, GroupK method uses a group of short k-mer hits satisfying statistically derived distance constraints to increase the sensitivity of small overlap detection.
Given the error profiles, such as the estimated indels and mismatch probabilities, thresholds for grouping short k-mers can be computed using the waiting time distribution and the one-dimensional random walk.
□ Dna-brnn: Identifying centromeric satellites
dna-brnn, a recurrent neural network to learn the sequences of the two classes of centromeric repeats. It achieves high similarity to RepeatMasker and is times faster. Dna-brnn explores a novel application of deep learning and may accelerate the study of the evolution of the two repeat classes of satellites.

□ MAPS: Model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006982
MAPS models the expected contact frequency of pairs of loci accounting for common biases of 3C methods, the PLAC-seq/HiChIP-specific biases and genomic distance effects, and uses this model to determine statistically significant long-range chromatin interactions.
MAPS adopts a zero-truncated Poisson regression framework to explicitly remove systematic biases in the PLAC-seq and HiChIP datasets, and then uses the normalized chromatin contact frequencies to identify significant chromatin interactions anchored at genomic regions bound.

□ OctConv: Reducing Spatial Redundancy in Convolutional Neural Networks with Octave Convolution
>> https://export.arxiv.org/pdf/1904.05049
OctConv is formulated as a single, generic, plug-and-play convolutional unit that can be used as a direct replacement of (vanilla) convolutions without any adjustments in the network architecture. An OctConv-equipped ResNet-152 can achieve 82.9% top-1 classification accuracy on ImageNet with merely 22.2 GFLOPs.

□ Multi-platform discovery of haplotype-resolved structural variation in human genomes
>> https://www.nature.com/articles/s41467-018-08148-z
a suite of long-read, short-read, strand-specific sequencing technologies, optical mapping, and variant discovery algorithms to comprehensively analyze three trios to define the full spectrum of human genetic variation in a haplotype-resolved manner.
using IL-based WGS should be analyzed using intersections of multiple SV-calling algorithms (Manta, Pindel, and Lumpy for deletion detection, and Manta and MELT for insertion detection) to gain a ~3% increase in sensitivity over individual methods while decreasing FDR from 7-3%.
□ TH-GRASP: accurate Prediction of Genome-wide RNA Secondary Structure Profile Based On Extreme Gradient Boosting
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/16/610782.full.pdf
a new method for end-to-end prediction of THe Genome-wide RNA Secondary Structure Profile (TH-GRASP) from RNA sequence by using the XGBoost. TH-GRASP was trained by using XGBoost, which is an ensemble method to generate k Classification and Regression Trees (CART).
