
LLMエージェントでDemand-Drivenな量子化学計算を実現

はじめに
CRSチームでソフトウェアエンジニアをやっている山口です!
こちらの記事は以下の記事の続きです。以下の記事からお読みいただくことをおすすめしますが知らなくても読めます。
前回は、Agentを利用してやりたいことベースで量子化学計算ができる「Demand-Drivenな量子化学計算」の全体像を解説しました。 今回は、どのようにしてやりたいことベースで量子化学計算するのかについて解説していきたいと思います。
やりたいことベースで量子化学計算を実行させるならAgentを使おう
やりたいことベースで量子化学計算を実行させる方法にはいくつかあります。
みなさんが最もよく知っているであろう機能の一つがAgentです。 "Agent"とは、モデルに意思決定の機能を与えて適切なアクションを選択・実行するシステムのことです。
やりたいことベースをリクエストとして入力して、一連のタスクを実行するプロセスは、以下のようになります
リクエストをエージェントに送る
リクエストに応じて、Agentがどのツールを使うとリクエストに答えられるかを推論する
そしてツールを使用する。
使用した結果から、リクエストに回答する。
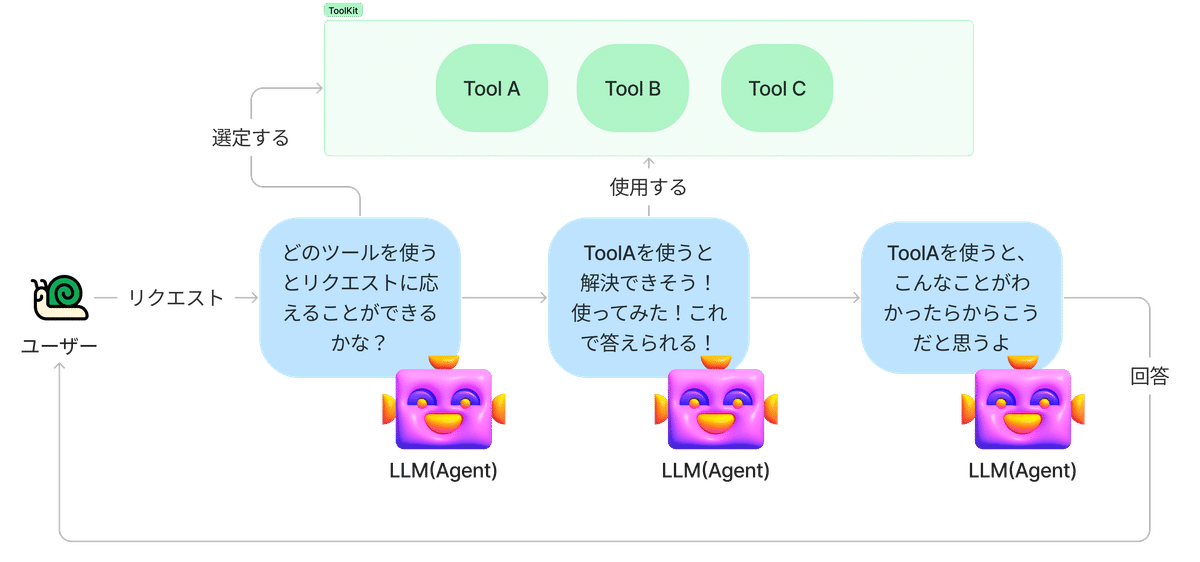
上記のプロセスでやりたいことベースで量子化学計算も実行できてしまうということです。
ではAgentについて詳しく説明していきましょう。
Agentとは?
Agentは自然言語処理と推論を組み合わせて、ユーザーからの指示を理解し、適切なアクションを選択・実行することで、タスクを自動的に完了させる仕組みです。
再度記載しますが、Agentとはモデルに意思決定の機能を与えて適切なアクションを選択・実行するシステムなんですね。
LangChain Agentの説明
LangChainはAgentの構築に必要な各種コンポーネントを提供し、開発者がAgentを容易に実装できるようにしています。
LangChainでのAgentのコンセプトは以下の図のようになっています。
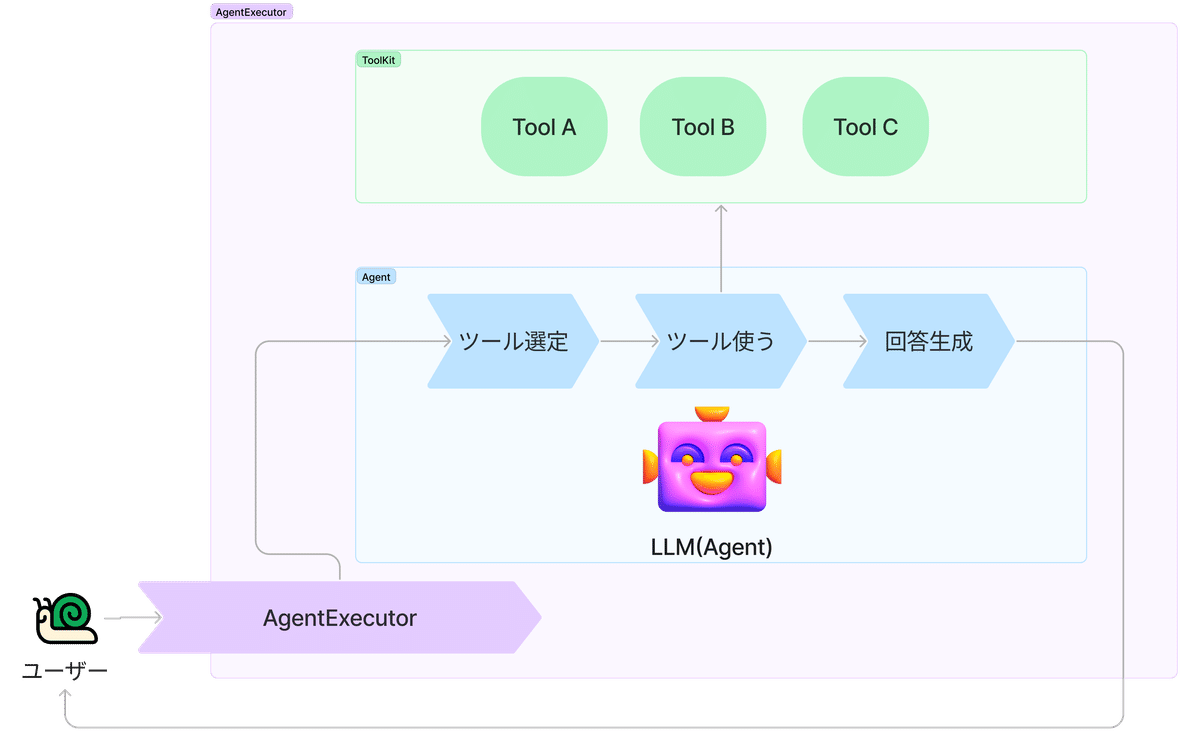
Agent
ユーザーからのリクエストに対して、持っているツールで何ができるかを考えて行動するもののこと。ここではLLMがその役割を担っています。
Tool
たとえば、「量子化学計算をするためのツール」、「arXivに論文を検索しにいくツール」、「chemRxivに論文に検索しにいくツール」などのことを言います。Agentがこのツールを使ってユーザーからのリクエストに対して行動します。
AgentExecutor
エージェントのランタイムです。これは、実際にエージェントを呼び出し、エージェントが選択したアクションを実行し、アクションの出力をエージェントに戻し、それを繰り返させることができます。
Agentを使用した具体例
Agentを使うことで、これまで人間が手作業で行っていたドメインに特化した専門的なタスクを、AIに自動化させることができます。
例えば、化学の分野で特定の化合物を計算するのに適した汎関数を探したい場合、Agentを使えば下記の図のように、目的の論文や汎関数を自動で見つけ出してくれます。
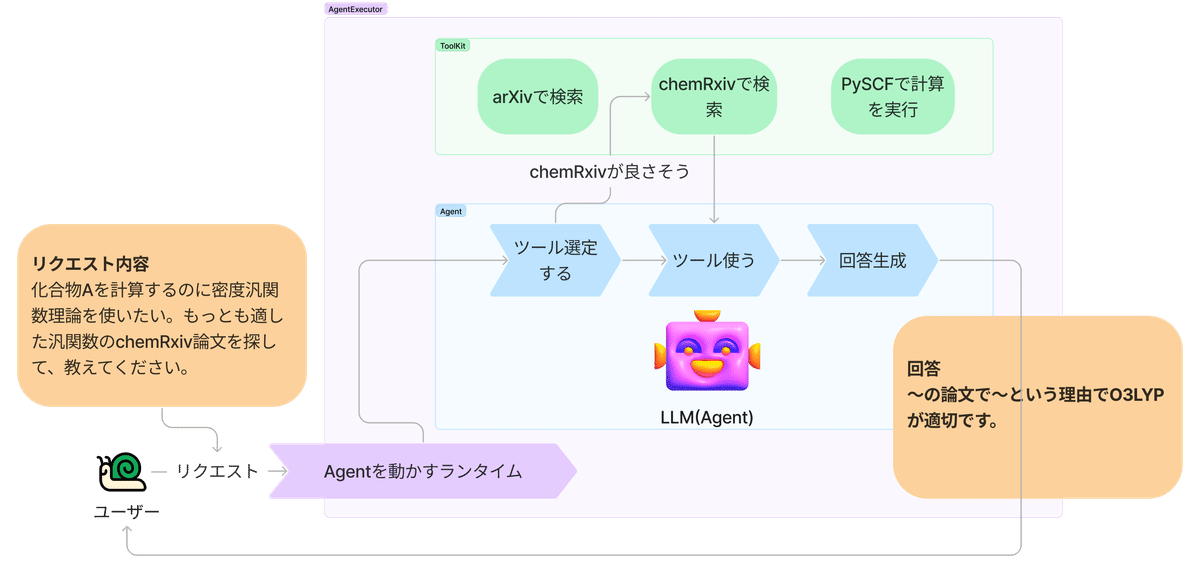
実際の例で解説していきましょう!
ユーザーリクエスト:化合物Aを計算するのに密度汎関数理論を使いたい。もっとも適した汎関数を探して、教えてください。
Agentの行動 :
1. 化合物A+DFT+functionalでchemRxiv検索
2. 情報取得してユーザーリクエストでベクトル検索
3. ユーザーリクエストに合わせて、Agentが最適な汎関数の情報を回答として生成
精度に関しては別途考える必要がありますが、Agentを使うことでこのようなことが可能になります。
実際に今回作成した「Demand-Drivenな量子化学計算」のシステムでも、このようなAgentの仕組みを利用しています。
ドメインに特化したタスクをLLMに実行させたい場合、Agent機能は必須の要素になってきます。
実装
では実際に実装にしてみましょう。
Python+LangChainを用いて実装します。
LLMの準備
AzureOpenAIを使用します。
import os
import openai
import getpass
from langchain_openai.chat_models import AzureChatOpenAI
# 環境変数の設定
os.environ["OPENAI_API_KEY"] = getpass.getpass()
openai.api_key = os.environ["OPENAI_API_KEY"]
azure_configs = {
"base_url": your-azure-base-url,
"model_deployment": your-model-deployment,
"model_name": "gpt-35-turbo-16k",
}
azure_model = AzureChatOpenAI(
openai_api_version="2023-05-15",
azure_endpoint=azure_configs["base_url"],
azure_deployment=azure_configs["model_deployment"],
model=azure_configs["model_name"],
validate_base_url=False,
)
ツールを用意
@toolをつけることでAgentが使うツールであることを明記します。以下を用意します。
- search_arxiv
- search_chemRxiv
- generate_molecule_from_smiles_and_calculate_energy
@tool
def search_arxiv(search_word: str):
"""Searches arXiv for tools"""
...実装省略
return result
@tool
def search_chemRxiv(search_word: str):
"""Searches chemRxiv for tools"""
...実装省略
return result
@tool
def generate_molecule_from_smiles_and_calculate_energy(smiles,functional:str="b3lyp"):
"""Generates a molecule from SMILES and calculates its energy using DFT"""
...実装省略
return result
tools = [search_arxiv, search_chemRxiv,generate_molecule_from_smiles_and_calculate_energy]
llm_with_tools = azure_model.bind(tools=[format_tool_to_openai_tool(tool) for tool in tools])エージェントを用意
エージェントを用意します。プロンプトは適宜用意してもらえるといいと思います。
prompt = ChatPromptTemplate.from_messages(
[
(
"system", "You are a very powerful assistant, especially in the field of quantum chemistry. "
"Talk with the user as normal. "
"If they ask you to calculate the energy of a molecule given its SMILES representation, use the generate_molecule_from_smiles_and_calculate_energy tool."
"Please let me know if this molecule is the right calculation for DFT after you have calculated it. If not suitable, what calculation should I do?Use select_general_function to select a general function"
),
MessagesPlaceholder(variable_name="chat_history"),
("user", "{input}"),
MessagesPlaceholder(variable_name="agent_scratchpad"),
]
)
agent = (
{
"input": lambda x: x["input"],
"agent_scratchpad": lambda x: format_to_openai_tool_messages(
x["intermediate_steps"]
),
"chat_history": lambda x: x["chat_history"],
}
| prompt
| llm_with_tools
| OpenAIToolsAgentOutputParser()
)エージェントの実行環境(Agent Executor)を用意
agentExecutor=AgentExecutor(agent=agent, tools=tools, verbose=True)実行する
agentExecutor.invoke("化合物Aを計算するのに密度汎関数理論を使いたい。もっとも適した汎関数のchemRxiv論文を探して、教えてください。")結果
結果は以下のようになります。長いのでみたい方は頑張って読んでください。
リクエスト内容:What is the best functional when doing a DFT calculation for benzene?
--
{'actions': [{'tool': 'search_chemrxiv', 'tool_input': {'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}, 'log': "\nInvoking: `search_chemrxiv` with `{'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_3DNPAjthgpi1vL3VyKQ9MGUW', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_3DNPAjthgpi1vL3VyKQ9MGUW'}], 'messages': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_3DNPAjthgpi1vL3VyKQ9MGUW', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})]}
--
{'steps': [{'action': {'tool': 'search_chemrxiv', 'tool_input': {'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}, 'log': "\nInvoking: `search_chemrxiv` with `{'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_3DNPAjthgpi1vL3VyKQ9MGUW', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_3DNPAjthgpi1vL3VyKQ9MGUW'}, 'observation': [Document(page_content='model against the complete analytical solution (Equation (12)), we leveraged MATLAB® \nR2022b; similar to [23], eigenvalues and eigenvectors of K were calculated using the eig \nfunction, nonlinear algebraic equations were solved implicitly using the fsolve function, and \nnumerical integration was performed using the cumtrapz function. The spreadsheet model used \nin this work was constructed in Microsoft® Excel® 2021 and leverages straightforward algebraic \nand matrix operations (i.e., mmult, transpose). All simulation results presented throughout this \nwork were performed on a Dell Latitude 7290 laptop computer with an Intel® Core™ i7-8650U \nprocessor (quad-core, 1.90 GHz) and a random-access memory of 16 GB. \n \n \nhttps://doi.org/10.26434/chemrxiv-2024-pvskd ORCID: https://orcid.org/0000-0001-6747-8197 Content not peer-reviewed by ChemRxiv. License: CC BY-NC-ND 4.0', metadata={'source': 'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf', 'file_path': 'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf', 'page': 13, 'total_pages': 37, 'format': 'PDF 1.4', 'title': '', 'author': '', 'subject': '', 'keywords': '', 'creator': 'Acrobat PDFMaker 23 for Word', 'producer': 'Adobe PDF Library 23.8.53', 'creationDate': "D:20240326130823-04'00'", 'modDate': "D:20240326130900-04'00'", 'trapped': ''}), Document(page_content='organometal halide perovskites. First Principles calculations were performed based on \nDFT calculation as implemented in the VASP program during which the spin-orbit effect \nwas neglected. These materials are direct bandgap material, which allow direct band to \nband transition with theoretically calculated tunable bandgap ranged 1.54 eV-2.33 eV \n(theoretically calculated) and 1.42-1.93 eV(experimentally determined). This suggests \nthat these materials are suitable for photovoltaic and other optoelectronic device \napplications such as laser, light emitting diode and photovoltaic devices, which require \ndirect band to band transitions. \n \nhttps://doi.org/10.26434/chemrxiv-2024-x8b26 ORCID: https://orcid.org/0000-0002-9961-4461 Content not peer-reviewed by ChemRxiv. License: CC BY 4.0', metadata={'source': 'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf', 'file_path': 'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf', 'page': 3, 'total_pages': 18, 'format': 'PDF 1.5', 'title': '', 'author': '', 'subject': '', 'keywords': '', 'creator': 'Microsoft® Word 2010', 'producer': 'Microsoft® Word 2010', 'creationDate': "D:20240329163953-07'00'", 'modDate': "D:20240329163953-07'00'", 'trapped': ''}), Document(page_content='9 \n \norbital components of each band can be seen more clearly in PDOS shown in Figure 3a-c. \nComplex bands between −3 and −1.5 eV are composed of I 5p and Cu 3d orbitals mixed \nslightly with Pb 6p and Cu 4s as hybridizing states. Furthermore, due to the hybridization \nof Cu 4s and X p orbitals the electron density at the Fermi energy is delocalized over the \nentire crystal. An important point is the existence of the bonding band of Cu 4s and I 5p \nabove the Fermi energy should govern the transport and conductivity properties of the \nsystem. Importantly, through the sequence CuPbCl3, CuPbBr3, and CuPbI3, the P and d \nlevels approach one another, causing an increase in the P-d mixing. In our model, an \nincreased mixing of P-d should decrease the width of the lower P band, a fact which is \nprobably compensated by the increased spin-orbit interaction in going from CuPbCl3 to \nCuPbI3. \nBecause of the antibonding nature of Pb—I26, the charge densities are localized at the', metadata={'source': 'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf', 'file_path': 'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf', 'page': 8, 'total_pages': 18, 'format': 'PDF 1.5', 'title': '', 'author': '', 'subject': '', 'keywords': '', 'creator': 'Microsoft® Word 2010', 'producer': 'Microsoft® Word 2010', 'creationDate': "D:20240329163953-07'00'", 'modDate': "D:20240329163953-07'00'", 'trapped': ''}), Document(page_content='Therefore, the charge time is the result of matrix operations corresponding to those species as \ngiven by Equation (15). \nhttps://doi.org/10.26434/chemrxiv-2024-pvskd ORCID: https://orcid.org/0000-0001-6747-8197 Content not peer-reviewed by ChemRxiv. License: CC BY-NC-ND 4.0', metadata={'source': 'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf', 'file_path': 'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf', 'page': 9, 'total_pages': 37, 'format': 'PDF 1.4', 'title': '', 'author': '', 'subject': '', 'keywords': '', 'creator': 'Acrobat PDFMaker 23 for Word', 'producer': 'Adobe PDF Library 23.8.53', 'creationDate': "D:20240326130823-04'00'", 'modDate': "D:20240326130900-04'00'", 'trapped': ''})]}], 'messages': [FunctionMessage(content='[Document(page_content=\'model against the complete analytical solution (Equation (12)), we leveraged MATLAB® \\nR2022b; similar to [23], eigenvalues and eigenvectors of K were calculated using the eig \\nfunction, nonlinear algebraic equations were solved implicitly using the fsolve function, and \\nnumerical integration was performed using the cumtrapz function. The spreadsheet model used \\nin this work was constructed in Microsoft® Excel® 2021 and leverages straightforward algebraic \\nand matrix operations (i.e., mmult, transpose). All simulation results presented throughout this \\nwork were performed on a Dell Latitude 7290 laptop computer with an Intel® Core™ i7-8650U \\nprocessor (quad-core, 1.90 GHz) and a random-access memory of 16 GB. \\n \\n \\nhttps://doi.org/10.26434/chemrxiv-2024-pvskd ORCID: https://orcid.org/0000-0001-6747-8197 Content not peer-reviewed by ChemRxiv. License: CC BY-NC-ND 4.0\', metadata={\'source\': \'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf\', \'file_path\': \'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf\', \'page\': 13, \'total_pages\': 37, \'format\': \'PDF 1.4\', \'title\': \'\', \'author\': \'\', \'subject\': \'\', \'keywords\': \'\', \'creator\': \'Acrobat PDFMaker 23 for Word\', \'producer\': \'Adobe PDF Library 23.8.53\', \'creationDate\': "D:20240326130823-04\'00\'", \'modDate\': "D:20240326130900-04\'00\'", \'trapped\': \'\'}), Document(page_content=\'organometal halide perovskites. First Principles calculations were performed based on \\nDFT calculation as implemented in the VASP program during which the spin-orbit effect \\nwas neglected. These materials are direct bandgap material, which allow direct band to \\nband transition with theoretically calculated tunable bandgap ranged 1.54 eV-2.33 eV \\n(theoretically calculated) and 1.42-1.93 eV(experimentally determined). This suggests \\nthat these materials are suitable for photovoltaic and other optoelectronic device \\napplications such as laser, light emitting diode and photovoltaic devices, which require \\ndirect band to band transitions. \\n \\nhttps://doi.org/10.26434/chemrxiv-2024-x8b26 ORCID: https://orcid.org/0000-0002-9961-4461 Content not peer-reviewed by ChemRxiv. License: CC BY 4.0\', metadata={\'source\': \'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf\', \'file_path\': \'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf\', \'page\': 3, \'total_pages\': 18, \'format\': \'PDF 1.5\', \'title\': \'\', \'author\': \'\', \'subject\': \'\', \'keywords\': \'\', \'creator\': \'Microsoft® Word 2010\', \'producer\': \'Microsoft® Word 2010\', \'creationDate\': "D:20240329163953-07\'00\'", \'modDate\': "D:20240329163953-07\'00\'", \'trapped\': \'\'}), Document(page_content=\'9 \\n \\norbital components of each band can be seen more clearly in PDOS shown in Figure 3a-c. \\nComplex bands between −3 and −1.5 eV are composed of I 5p and Cu 3d orbitals mixed \\nslightly with Pb 6p and Cu 4s as hybridizing states. Furthermore, due to the hybridization \\nof Cu 4s and X p orbitals the electron density at the Fermi energy is delocalized over the \\nentire crystal. An important point is the existence of the bonding band of Cu 4s and I 5p \\nabove the Fermi energy should govern the transport and conductivity properties of the \\nsystem. Importantly, through the sequence CuPbCl3, CuPbBr3, and CuPbI3, the P and d \\nlevels approach one another, causing an increase in the P-d mixing. In our model, an \\nincreased mixing of P-d should decrease the width of the lower P band, a fact which is \\nprobably compensated by the increased spin-orbit interaction in going from CuPbCl3 to \\nCuPbI3. \\nBecause of the antibonding nature of Pb—I26, the charge densities are localized at the\', metadata={\'source\': \'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf\', \'file_path\': \'CuPbX3 and AgPbX3 Inorganic Perovskites for Solar cell Applications .pdf\', \'page\': 8, \'total_pages\': 18, \'format\': \'PDF 1.5\', \'title\': \'\', \'author\': \'\', \'subject\': \'\', \'keywords\': \'\', \'creator\': \'Microsoft® Word 2010\', \'producer\': \'Microsoft® Word 2010\', \'creationDate\': "D:20240329163953-07\'00\'", \'modDate\': "D:20240329163953-07\'00\'", \'trapped\': \'\'}), Document(page_content=\'Therefore, the charge time is the result of matrix operations corresponding to those species as \\ngiven by Equation (15). \\nhttps://doi.org/10.26434/chemrxiv-2024-pvskd ORCID: https://orcid.org/0000-0001-6747-8197 Content not peer-reviewed by ChemRxiv. License: CC BY-NC-ND 4.0\', metadata={\'source\': \'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf\', \'file_path\': \'A spreadsheet-based redox flow battery cell cycling model enabled by closed-form approximations.pdf\', \'page\': 9, \'total_pages\': 37, \'format\': \'PDF 1.4\', \'title\': \'\', \'author\': \'\', \'subject\': \'\', \'keywords\': \'\', \'creator\': \'Acrobat PDFMaker 23 for Word\', \'producer\': \'Adobe PDF Library 23.8.53\', \'creationDate\': "D:20240326130823-04\'00\'", \'modDate\': "D:20240326130900-04\'00\'", \'trapped\': \'\'})]', name='search_chemrxiv')]}
--
{'output': 'The search did not directly provide information on the best functional for DFT calculations specifically for benzene. It seems the search results were not focused on benzene or its computational chemistry aspects, specifically concerning the choice of functionals in DFT calculations.\n\nFor DFT calculations of benzene or similar organic molecules, commonly used functionals include B3LYP, which is a hybrid functional combining Hartree-Fock exchange with gradient-corrected correlation, and PBE (Perdew-Burke-Ernzerhof), a generalized gradient approximation (GGA) functional. The choice between these or other functionals often depends on the specific properties you are interested in and the level of accuracy required for your study.\n\nFor accurate predictions of properties such as electronic structure, reaction energies, or vibrational frequencies, B3LYP is widely used and often provides a good balance between computational cost and accuracy. However, for certain properties or systems, other functionals might offer better performance.\n\nIf you are conducting research on benzene or planning a computational study, it may be beneficial to consult recent literature on quantum chemistry calculations for benzene to see which functionals have been recommended or used successfully for similar studies.', 'messages': [AIMessage(content='The search did not directly provide information on the best functional for DFT calculations specifically for benzene. It seems the search results were not focused on benzene or its computational chemistry aspects, specifically concerning the choice of functionals in DFT calculations.\n\nFor DFT calculations of benzene or similar organic molecules, commonly used functionals include B3LYP, which is a hybrid functional combining Hartree-Fock exchange with gradient-corrected correlation, and PBE (Perdew-Burke-Ernzerhof), a generalized gradient approximation (GGA) functional. The choice between these or other functionals often depends on the specific properties you are interested in and the level of accuracy required for your study.\n\nFor accurate predictions of properties such as electronic structure, reaction energies, or vibrational frequencies, B3LYP is widely used and often provides a good balance between computational cost and accuracy. However, for certain properties or systems, other functionals might offer better performance.\n\nIf you are conducting research on benzene or planning a computational study, it may be beneficial to consult recent literature on quantum chemistry calculations for benzene to see which functionals have been recommended or used successfully for similar studies.')]}結果を簡単にまとめると、
「うまく適切な値を見つけられなかったので、有機化合物でよく使われるB3LYPとかPBEなどの汎関数がいいんじゃないですか?有用なものが他の文献にあるかもしれないからそっちみて」ということです。
ドキュメントから見つからなかった場合はかなり煮え切らない回答になりました。もっとはっきりと汎関数を回答してもらいたいです。
少し修正した結果
ドキュメントから見つからなかった時もはっきりと汎関数を選択して欲しいのです。
そこで、以下のようなツールを介することで、LLMが汎関数の選択をより適切に行えるようにしました。
以下のコードで、汎関数とその選択理由を生成します。
def select_general_function(query: str,context:Document[] ):
"""Selects a general function based on the condition"""
class DFTFunctional(BaseModel):
functional: Literal[
"SLATERX", "PW86X", "VWN3C", "VWN5C", "PBEC", "PBEX", "BECKEX", "BECKECORRX", "BECKESRX", "BECKECAMX", "BRX", "BRC", "BRXC", "LDAERFX", "LDAERFC", "LDAERFC_JT", "LYPC", "OPTX", "OPTXCORR", "REVPBEX", "RPBEX", "SPBEC", "VWN_PBEC", "KTX", "TFK", "TW", "PW91X", "PW91K", "PW92C", "M05X", "M05X2X", "M06X", "M06X2X", "M06LX", "M06HFX", "M05X2C", "M05C", "M06C", "M06HFC", "M06LC", "M06X2C", "TPSSC", "TPSSX", "REVTPSSC", "REVTPSSX", "SCANC", "SCANX", "RSCANC", "RSCANX", "RPPSCANC", "RPPSCANX", "R2SCANC", "R2SCANX", "R4SCANC", "R4SCANX", "PZ81C", "P86C", "P86CORRC", "BTK", "VWK", "B97X", "B97C", "B97_1X", "B97_1C", "B97_2X", "B97_2C", "CSC", "APBEC", "APBEX", "ZVPBESOLC", "PBEINTC", "PBEINTX", "PBELOCC", "PBESOLX", "TPSSLOCC", "ZVPBEINTC", "PW91C", "SLATER", "LDA", "VWN", "VWN5", "VWN3", "SVWN", "B88", "LYP", "P86", "M052XX", "M062XX", "M052XC", "M062XC", "BLYP", "BP86", "BPW91", "BPW92", "OLYP", "KT1X", "KT2XC", "KT3XC", "PBE0", "PBE1PBE", "PBEH", "B3P86", "B3P86G", "B3P86V5", "B3PW91", "B3LYP", "B3LYP5", "B3LYPG", "O3LYP", "X3LYP", "X3LYPG", "X3LYP5", "CAMB3LYP", "CAM_B3LYP", "LDAERF", "B97XC", "B97_1XC", "B97_2XC", "TPSSH", "TF", "B97M_V", "WB97M_V", "WB97X_V", "VV10", "LC_VV10", "REVSCAN_VV10", "SCAN_RVV10", "SCAN_VV10", "SCANL_RVV10", "SCANL_VV10", "B97MV", "WB97MV", "WB97XV", "LCVV10", "REVSCANVV10", "SCANRVV10", "SCANVV10", "SCANLRVV10", "SCANLVV10"
] = Field(description="The chosen DFT functional for the calculation")
reason: str = Field(None, description="The reason for choosing the functional")
@validator("functional")
def validate_functional(cls, field):
# This validator is not strictly necessary as the Literal type will enforce the allowed values.
# However, you could add additional logic here if needed.
return field
# Set up a parser for DFT functional
parser = PydanticOutputParser(pydantic_object=DFTFunctional)
prompt = PromptTemplate(
template="""You are a very powerful assistant, especially in the field of quantum chemistry.
You has a deep understanding of the principles of quantum mechanics and computational chemistry, enabling them to expertly select the most appropriate functional for DFT calculations based on the specific requirements of each research project.
Please select an output format from the following
[SLATERX, PW86X, VWN3C, VWN5C, PBEC, PBEX, BECKEX, BECKECORRX, BECKESRX, BECKECAMX, BRX, BRC, BRXC, LDAERFX, LDAERFC, LDAERFC_JT, LYPC, OPTX, OPTXCORR, REVPBEX, RPBEX, SPBEC, VWN_PBEC, KTX, TFK, TW, PW91X, PW91K, PW92C, M05X, M05X2X, M06X, M06X2X, M06LX, M06HFX, M05X2C, M05C, M06C, M06HFC, M06LC, M06X2C, TPSSC, TPSSX, REVTPSSC, REVTPSSX, SCANC, SCANX, RSCANC, RSCANX, RPPSCANC, RPPSCANX, R2SCANC, R2SCANX, R4SCANC, R4SCANX, PZ81C, P86C, P86CORRC, BTK, VWK, B97X, B97C, B97_1X, B97_1C, B97_2X, B97_2C, CSC, APBEC, APBEX, ZVPBESOLC, PBEINTC, PBEINTX, PBELOCC, PBESOLX, TPSSLOCC, ZVPBEINTC, PW91C, SLATER, LDA, VWN, VWN5, VWN3, SVWN, B88, LYP, P86, M052XX, M062XX, M052XC, M062XC, BLYP, BP86, BPW91, BPW92, OLYP, KT1X, KT2XC, KT3XC, PBE0, PBE1PBE, PBEH, B3P86, B3P86G, B3P86V5, B3PW91, B3LYP, B3LYP5, B3LYPG, O3LYP, X3LYP, X3LYPG, X3LYP5, CAMB3LYP, CAM_B3LYP, LDAERF, B97XC, B97_1XC, B97_2XC, TPSSH, TF, B97M_V, WB97M_V, WB97X_V, VV10, LC_VV10, REVSCAN_VV10, SCAN_RVV10, SCAN_VV10, SCANL_RVV10, SCANL_VV10, B97MV, WB97MV, WB97XV, LCVV10, REVSCANVV10, SCANRVV10, SCANVV10, SCANLRVV10, SCANLVV10]
And tell me why you chose that generality.
.\n{format_instructions}\n{query}\n{context}""",
input_variables=["query"],
partial_variables={"format_instructions": parser.get_format_instructions()},
)
chain = prompt | azure_model | parser
result = chain.invoke({"query": query, "context":context })
functional = result.functional
reason = result.reason
print(f"Selected functional: {functional}")
return {
"functional": functional,
"reason": reason
}では、上記のコードを踏まえて実行してみましょう。
リクエスト内容:What is the best functional when doing a DFT calculation for benzene?
--
{'actions': [{'tool': 'search_chemrxiv_and_select_functional', 'tool_input': {'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}, 'log': "\nInvoking: `search_chemrxiv_and_select_functional` with `{'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_31eSZX4jS2JNJ4a36MQWEbjf', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_31eSZX4jS2JNJ4a36MQWEbjf'}], 'messages': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_31eSZX4jS2JNJ4a36MQWEbjf', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})]}
--
{'steps': [{'action': {'tool': 'search_chemrxiv_and_select_functional', 'tool_input': {'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}, 'log': "\nInvoking: `search_chemrxiv_and_select_functional` with `{'search_keywords': 'benzene DFT functional', 'query': 'What is the best functional when doing a DFT calculation for benzene?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_31eSZX4jS2JNJ4a36MQWEbjf', 'function': {'arguments': '{"search_keywords":"benzene DFT functional","query":"What is the best functional when doing a DFT calculation for benzene?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_31eSZX4jS2JNJ4a36MQWEbjf'}, 'observation': {'functional': 'B3LYP', 'reason': 'B3LYP is chosen for its balanced treatment of exchange and correlation effects, making it suitable for accurately predicting the properties of organic molecules like benzene. It is a hybrid functional that is widely used and trusted in the quantum chemistry community for a variety of applications, offering a good compromise between computational cost and accuracy.'}}], 'messages': [FunctionMessage(content='{"functional": "B3LYP", "reason": "B3LYP is chosen for its balanced treatment of exchange and correlation effects, making it suitable for accurately predicting the properties of organic molecules like benzene. It is a hybrid functional that is widely used and trusted in the quantum chemistry community for a variety of applications, offering a good compromise between computational cost and accuracy."}', name='search_chemrxiv_and_select_functional')]}
--
{'output': 'The best functional for doing a DFT calculation for benzene is B3LYP. This functional is chosen for its balanced treatment of exchange and correlation effects, making it suitable for accurately predicting the properties of organic molecules like benzene. B3LYP is a hybrid functional that is widely used and trusted in the quantum chemistry community for a variety of applications, offering a good compromise between computational cost and accuracy.', 'messages': [AIMessage(content='The best functional for doing a DFT calculation for benzene is B3LYP. This functional is chosen for its balanced treatment of exchange and correlation effects, making it suitable for accurately predicting the properties of organic molecules like benzene. B3LYP is a hybrid functional that is widely used and trusted in the quantum chemistry community for a variety of applications, offering a good compromise between computational cost and accuracy.')]}自信持って「B3LYPを使え!」って言ってくれますね!
ベンゼンみたいな有機化合物に対して汎函数B3LYPはよく使われているのでエージェントを使った意味はあまり見出せませんね。
ではB3LYP以外の回答が出る可能性のある金属が入った 「Co(C5H5)2」で試してみましょう。
リクエスト内容:What is the best functional when doing a DFT calculation for Co(C5H5)2?
--
{'actions': [{'tool': 'search_chemrxiv_and_select_functional', 'tool_input': {'search_keywords': 'DFT functional for Co(C5H5)2', 'query': 'What is the best functional when doing a DFT calculation for Co(C5H5)2?'}, 'log': "\nInvoking: `search_chemrxiv_and_select_functional` with `{'search_keywords': 'DFT functional for Co(C5H5)2', 'query': 'What is the best functional when doing a DFT calculation for Co(C5H5)2?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_OgfUk2YkTzVYM1QwNLJor1eR', 'function': {'arguments': '{"search_keywords":"DFT functional for Co(C5H5)2","query":"What is the best functional when doing a DFT calculation for Co(C5H5)2?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_OgfUk2YkTzVYM1QwNLJor1eR'}], 'messages': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_OgfUk2YkTzVYM1QwNLJor1eR', 'function': {'arguments': '{"search_keywords":"DFT functional for Co(C5H5)2","query":"What is the best functional when doing a DFT calculation for Co(C5H5)2?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})]}
--
{'steps': [{'action': {'tool': 'search_chemrxiv_and_select_functional', 'tool_input': {'search_keywords': 'DFT functional for Co(C5H5)2', 'query': 'What is the best functional when doing a DFT calculation for Co(C5H5)2?'}, 'log': "\nInvoking: `search_chemrxiv_and_select_functional` with `{'search_keywords': 'DFT functional for Co(C5H5)2', 'query': 'What is the best functional when doing a DFT calculation for Co(C5H5)2?'}`\n\n\n", 'type': 'AgentActionMessageLog', 'message_log': [AIMessageChunk(content='', additional_kwargs={'tool_calls': [{'index': 0, 'id': 'call_OgfUk2YkTzVYM1QwNLJor1eR', 'function': {'arguments': '{"search_keywords":"DFT functional for Co(C5H5)2","query":"What is the best functional when doing a DFT calculation for Co(C5H5)2?"}', 'name': 'search_chemrxiv_and_select_functional'}, 'type': 'function'}]}, response_metadata={'finish_reason': 'tool_calls'})], 'tool_call_id': 'call_OgfUk2YkTzVYM1QwNLJor1eR'}, 'observation': {'functional': 'M06LX', 'reason': 'M06L (Minnesota 2006 Local) is chosen for its balanced treatment of exchange and correlation effects, particularly in transition metal complexes, where it accurately captures the strong correlation effects and potential multi-reference character without the computational expense of non-local exchange.'}}], 'messages': [FunctionMessage(content='{"functional": "M06LX", "reason": "M06L (Minnesota 2006 Local) is chosen for its balanced treatment of exchange and correlation effects, particularly in transition metal complexes, where it accurately captures the strong correlation effects and potential multi-reference character without the computational expense of non-local exchange."}', name='search_chemrxiv_and_select_functional')]}
--
{'output': 'The best functional for doing a DFT calculation for Co(C5H5)2 is M06L (Minnesota 2006 Local, also referred to as M06LX in the response). This functional is chosen for its balanced treatment of exchange and correlation effects, particularly in transition metal complexes like Co(C5H5)2. It accurately captures the strong correlation effects and potential multi-reference character without the computational expense of non-local exchange. This makes it suitable for dealing with the complexities associated with transition metals.', 'messages': [AIMessage(content='The best functional for doing a DFT calculation for Co(C5H5)2 is M06L (Minnesota 2006 Local, also referred to as M06LX in the response). This functional is chosen for its balanced treatment of exchange and correlation effects, particularly in transition metal complexes like Co(C5H5)2. It accurately captures the strong correlation effects and potential multi-reference character without the computational expense of non-local exchange. This makes it suitable for dealing with the complexities associated with transition metals.')]}
遷移金属錯体なので、M06Lを選択してますね。M06系を選んでいるのでまあいいんじゃないでしょうか?
ただこれも文献は見つかってないようですので、LLMが考えて出してくれた回答のようです。
文献見つからない時の対処法
文献が見つかるようにするには以下が対処法になりそうです。
検索の媒体を変える。
自社で知見が溜まっているなら、それも使えます。
他の特定のジャーナルなどもRAGとして使う方法もあります。
検索ワードを変更する。
LangChain Agentの問題点
Agentの機能についてはいかがでしたでしょうか?
ドメインに特化した様々なユースケースが思い浮かぶかもしれません。
ただ、このままでは前回の記事のデモで見ていただいたような柔軟性のあるシステムにはならないのです。
例えば以下のようなことはできません。
「ツールを実行したけど、ユーザーのリクエストに答えられなさそうだから別のツールで実行する」
「まだこの構造は最適化されていないからもう一度最適化させるべきか
なぜならLLM Agent自体は、LangChainのLCELに沿っているため、試行サイクルを繰り返すような処理や複数のエージェントを使うようなことに関しては向いていないからです。
そんなときに使用できるのが以下のようなマルチエージェントツールです。
これらを使うとLangChain Agent単体できなかったことが解消できます。
柔軟なAgentシステムを実装するなら?
それぞれAgentを使うフレームワークとしては良さがありますが、Demand-Drivenな量子化学計算ではLangGraphを使用しています。
LangGraphを使う良さ
LangGraphは、Agentをより柔軟に活用するための強力なツールです。LangGraphの主な特徴は、LangChainでは扱えなかった閉路(サイクル)を含むグラフ構造の処理を可能にしたことです。
ですが、それだけが利点ではありません。
グラフを自分で考えるのでシンプルで実装の見通しが立てやすく使いやすい
LangGraphではグラフ構造を自分で設計するため、処理の流れが明確になります。これにより、実装の見通しが立てやすくなり、使いやすさが向上します。
試行サイクルを回せるので、LangChain Agentと相性がいい
LangGraphを使うことで、LangChain Agentに考えさせた結果を見て、さらにブラッシュアップさせるといった試行サイクルを回すことが容易になります。これにより、Agentの能力を最大限に引き出すことができます。
各ドメインに特化させたAgentを複数用意し、行動させて試行サイクルを回すなんてこともできます。
特に、複雑なAgentシステムを構築する際には、LangGraphを使うことで開発の効率化と品質向上が期待できると思います。
もし複雑なAgentでのシステムを作成する時はLangGraphを使ってみてください!
最後に
Agentの使い方についてはいかがでしたでしょうか?
もしAgentを使ったユースケースが思いつきましたら、ぜひ実装チャレンジしてみてください!
LLM✖︎化学領域でお困りのことがある方がいましたらお気軽に下記までお問い合わせください!