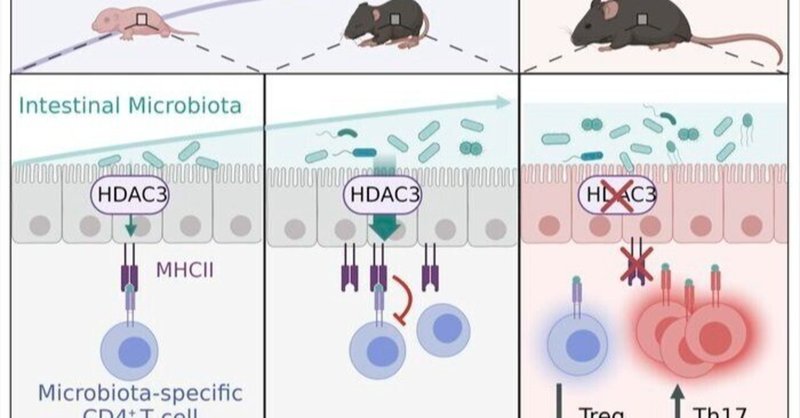
腸管上皮のHDAC3とMHCクラスIIは微生物特異的免疫を調整する

研究論文消化器免疫学オープンアクセス|10.1172/JCI162190
腸管上皮のHDAC3とMHCクラスIIは微生物特異的免疫を調整する
https://www.jci.org/articles/view/162190
Emily M. Eshleman,1,2 Tzu-Yu Shao,2,3,4 Vivienne Woo,1,2 Taylor Rice,1,2 Laura Engleman,1,2 Bailey J. Didriksen,1,2,4 Jordan Whitt,1,2 David B. Haslam,3 Sing Sing Way,2,3 and Theresa Alenghat1,2
2023年1月5日発行 - 詳細はこちら
PDFを見る
要旨
常在微生物に対する異常な免疫応答は、炎症性腸疾患やその他の慢性炎症状態を促進する。しかし、粘膜組織において微生物特異的免疫がどのように制御されているかは、まだ十分に理解されていない。ここで我々は、微生物叢に感受性を示すヒストン脱アセチル化酵素3(HDAC3)の上皮発現を欠損したマウスでは、腸内の常在細菌特異的CD4+ T細胞の蓄積が増加することを見いだし、上皮HDAC3が局所の微生物叢特異的免疫に指示を与えているのではないかという仮説を提唱した。これと一致して、微生物特異的CD4+ T細胞と上皮性HDAC3の発現は、早期の微生物叢コロニー形成後に同時に誘導された。さらに、HDAC3の上皮内在性切除は、常在菌特異的Tregを減少させ、常在菌特異的Th17細胞を増加させ、T細胞駆動性大腸炎を促進した。メカニズム的には、HDAC3はNF-κB依存的な上皮性MHCクラスII(MHCII)の制御に必須であった。上皮内在性MHCIIは、成体マウスにおいて、常在菌特異的Th17細胞の局所的蓄積を抑制し、微生物が誘発する炎症から保護した。驚くべきことに、HDAC3は、微生物叢が上皮細胞上でMHCIIの発現を誘導し、腸内の常在菌特異的T細胞の数を制限することを可能にした。これらのデータを総合すると、常在細菌を認識し、炎症を制御する組織内在性のCD4+ T細胞サブセットの動的バランスを制御する上で、上皮ヒストン脱アセチル化酵素が中心的な役割を担っていることが明らかになった。

図解抄録
はじめに
消化管には、微生物叢と総称される数兆個もの微生物が生息しており、これらの微生物は哺乳動物細胞と共生関係を形成し、健康と疾病を媒介する上で重要な役割を果たしている。広域抗生物質や無菌マウスモデルを用いた広範な実験により、宿主免疫系の発達と機能において微生物叢が必要であることが明らかになっている(1-5)。微生物叢の相互作用は、生後数年の間に重要な免疫教育や較正が行われるため、生後間もない時期に特に大きな影響を及ぼす(6-9)。実際、この生後間もない時期における微生物叢の構成やコロニー形成の乱れや擾乱は、喘息、アレルギー、炎症性腸疾患(IBD)を含む慢性炎症性疾患の発症の素因となりうる免疫の長期にわたる変化と関連している(1, 4, 8)。
宿主と微生物叢の関係は共生的であるにもかかわらず、粘膜表面に抗原的に外来微生物が多く存在し、密接に関連しているため、病的炎症を刺激する潜在的リスクがある。このため、腸管免疫応答は、侵入してくる病原体に対する防御免疫を可能にする一方で、無害な常在微生物に対する炎症反応を制限するように、厳密に制御されなければならない。常在細菌は制御性T細胞の分化を促進し(10)、微生物に反応するエフェクターT細胞とメモリーT細胞がマウスとヒトの両方に存在する(11-17)。これらの常在菌特異的T細胞は、防御サイトカインを誘導し、病原体に対する交差反応性を提供することにより、バリア機能を促進する(14, 18, 19)。しかし、微生物叢に対する異常な免疫応答は、IBDのような炎症状態の引き金にもなる(1, 20-22)。大腸炎のマウスモデルでは、腸内細菌は、部分的には微生物叢に反応するCD4+ T細胞を刺激することによって炎症を引き起こす(23-25)。さらに、IBD患者の微生物特異的CD4+ T細胞は、健常患者のT細胞と比較して、機能的に変化しており、IL-17などの炎症性サイトカインをより多く産生することが示されている(14, 26-30)。しかし、常在菌特異的CD4+ T細胞の組織内制御を指示するメカニズムは、まだ十分に解明されていない。
腸管上皮細胞(IEC)は、微生物叢と免疫細胞との直接の接点に存在するため、微生物抗原に応答する局所免疫を指示する独自の態勢を整えている。IECは病原体認識レセプターを発現しており、微生物のシグナルを感知し、サイトカイン、ケモカイン、成長因子の分泌を介して腸管免疫応答を制御する(1, 31)。上皮内の特殊な小葉細胞や杯細胞関連抗原通路が抗原を下層の抗原提示ミエロイド細胞に送り込む一方で(32-34)、IECは免疫応答を制御しうる古典的な抗原処理・提示経路も備えている(35, 36)。しかし、健全な微生物と免疫の関係を調整するIEC主導のメカニズムについては、まだ理解が不十分である。
正統的なセンサー以外にも、エピジェネティック修飾酵素であるヒストン脱アセチル化酵素3(HDAC3)の上皮発現が微生物叢に応答し、哺乳類の代謝、腸内恒常性、炎症を制御していることが最近わかってきた(37-40)。ここで我々は、最初の微生物叢コロニー形成後の常在菌特異的CD4+ T細胞の微生物叢依存的な増殖は、HDAC3の上皮発現と同時に起こることを見出した。この関係を踏まえて、我々はIEC内在性のHDAC3が、微生物叢に刺激されたCD4+ T細胞の増殖と分化を制御しているかどうかを調べた。実際、IECにおけるHDAC3発現の消失は、腸の炎症と、上皮性MHCIIによって誘導される微生物特異的Th17細胞の蓄積をもたらした。無菌条件下で飼育したマウスの組織をさらに分析したところ、微生物特異的CD4+ T細胞の上皮性MHCII依存的制御を微生物叢が誘導するためには、HDAC3が必要であることが示された。これらのデータを総合すると、微生物叢は、組織内在性T細胞の上皮制御を指示することによって、常在性寛容を誘導し、炎症を制限することが明らかになった。
研究結果
上皮性HDAC3発現は腸内常在菌特異的CD4+ T細胞を制限する。微生物叢のコロニー形成は出生時に始まり、微生物叢の複雑さと密度は乳児期に最も顕著に増加する(41-43)。生後間もない時期から微生物叢にさらされると免疫教育が促進され、この時期に微生物叢が破壊されると慢性炎症性疾患が増加する可能性がある(42-46)。出生時の初期コロニー形成は、常在微生物に対する特異性を持つT細胞の蓄積を促すと予測されているが、このことは直接検証されていない。そこで、生後早期の微生物叢のコロニー形成そのものが、腸内の常在細菌特異的T細胞応答を誘導するのかどうかを調べるために、無菌(GF)と通常飼育(CNV)の新生仔の大腸から採取したCD4+ T細胞を用いて、まずMHCII制限四量体を用いて、常在べん毛cBir1ペプチドに対するT細胞レセプター特異性を解析した。生後1週間以内に、GFおよびCNVの仔マウスは、大腸における常在菌特異的cBir1+CD4+ T細胞の存在量が同様に少なかった(図1、AおよびB)。対照的に、微生物存在下で飼育された3週齢の仔マウスは、年齢をマッチさせたGF対照と比較してcBir1+CD4+ T細胞の蓄積が増加した(図1、AおよびB)。これは、離乳までに微生物叢のコロニー形成が徐々に増加するのと類似している(44)。したがって、微生物叢由来のシグナルは、腸内細菌特異的CD4+ T細胞の初期増殖のプライミングに不可欠である。
図1
上皮性HDAC3発現は腸内常在菌特異的CD4+ T細胞を制限する。(AおよびB)新生児GFおよびCNV仔の大腸から単離したcBir1+四量体特異的CD4+ T細胞の数。 C)GFおよびCNV仔の大腸から単離したIECにおけるHDAC3のmRNA発現。 DおよびE)全腸CD3+の頻度。(FおよびG)腸管CD8a+(F)およびCD4+(G)T細胞の頻度。(H-K) HDAC3FFおよびHDAC3ΔIECマウスの大腸におけるcBir1+四量体特異的CD4+ T細胞の数(HおよびI)、およびRORγt+(J)およびFoxP3+(K)cBir1+CD4+ T細胞の頻度。cBir1+四量体細胞は、生細胞、CD45+、系統(CD11b-B220-Ly6G-、CD11c-CD8a-、CD4+)でゲートされる。データは少なくとも2回の実験、1群3-4匹のマウスの代表値。*P<0.05、***P<0.01、***P<0.0001、Tukeyの多重比較検定による1-way ANOVA(B)、または対応のない2-tailed Studentのt検定(C-K)。
制御不全の常在特異的T細胞は腸の炎症と関連しており(14)、これらのT細胞サブセットを制御する経路の障害が、病的炎症への感受性に影響を及ぼす可能性が示唆される。興味深いことに、IBD患者のIECはHDAC3酵素の発現レベルが低下しており(37)、他の施設での知見と一致して、HDAC3のIEC内在性発現を欠損したマウス(HDAC3ΔIECマウス)は、直腸脱を特徴とする慢性腸炎に対する感受性が上昇していた(補足図1A;本論文とともにオンラインで入手可能な補足資料;https://doi. org/10.1172/JCI162190DS1)、炎症性バイオマーカーであるリポカリン-2のレベル上昇(補足図1B)、炎症性細胞の浸潤(補足図1、C-E)、慢性炎症と一致する組織学的変化(補足図1F)を示した(37)。驚くべきことに、HDAC3の上皮発現もまた、1週齢の仔と比較して3週齢の仔で劇的に誘導された(図1C)。しかし、GF条件下で飼育された仔マウスでは、上皮性HDAC3の発現はこれらの発育ウィンドウにおいてバックグラウンドレベルの低いままであった(図1C)。このことは、微生物叢のコロニー形成が腸の初期上皮性HDAC3発現を増加させることを示している。
微生物叢コロニー形成後の上皮内在性HDAC3発現と常在菌特異的CD4+ T細胞の拡大との間の時間的関連は、IEC内在性HDAC3が微生物特異的T細胞免疫を制御しているのではないかという仮説を引き起こした。腸管固有層では、HDAC3ΔIECマウスでは、Cre陰性同腹のHDAC3FFコントロールと比較して、CD3+ T細胞の総数は変化していなかった(図1、DおよびE)。しかしながら、上皮性HDAC3の欠損は、大腸(図1、HおよびI)および小腸(補足図2、AおよびB)においてcBir1+常在特異性の増加を示したが、CD8a+細胞ではなく、腸CD4+ T細胞の上昇をもたらした(図1、FおよびG)。メタゲノム解析の結果、HDAC3ΔIECマウスの大腸における常在菌の組成が変化していることが確認され(補足図3A)、多様性の減少(補足図3B)とビフィズス菌の減少(補足図3C)が特徴的であり、先行する16S配列決定(37)と一致した。さらに、HDAC3ΔIECマウスの腸内細菌叢では、cBir1の発現自体が、同腹のコントロールに比べて減少していた(補足図3、DおよびE)。したがって、HDAC3ΔIECマウスにおけるcBir1-間質特異的T細胞の増加は、cBir1発現種のレベルとは並行していない。HDAC3ΔIECマウスにおけるcBir1+ CD4+T細胞は、炎症性RORγt+ Th17細胞への分化が増加し(図1J)、FoxP3+ T制御性分化は相互に減少した(図1K)。日和見特異的Th1とT濾胞ヘルパー(Tfh)細胞は、上皮HDAC3の欠損によって変化しなかった(補足図4A)。これらのデータを総合すると、上皮性HDAC3は腸内の常在菌特異的TregとTh17細胞のバランスを制御する上で重要な役割を果たしていることがわかる。
上皮性HDAC3を欠損したマウスのCD4+ T細胞は、重篤な大腸炎を誘発する。HDAC3ΔIECマウスの調節不全CD4+ T細胞が腸炎を促進するかどうかを調べるために、慢性大腸炎のT細胞移入モデル(47-49)を用いた。このモデルでは、HDAC3FFマウスおよびHDAC3ΔIECマウスから精製CD4+ T細胞を単離し、Rag1-/-レシピエントマウスに養子移入した(図2A)。対照マウスからCD4+ T細胞を受け取ったRag1-/-レシピエントとは対照的に、HDAC3ΔIECマウスからCD4+ T細胞を受け取ったRag1-/-レシピエントは、より顕著な体重減少(図2B)、結腸短縮(図2C)、および炎症細胞浸潤、陰窩過形成、壁肥厚を特徴とする重篤な大腸炎病態を示した(図2、DおよびE)。さらに、HDAC3ΔIECマウスからCD4+T細胞を受け取ったRag1-/-マウスでは、炎症性バイオマーカーであるリポカリン-2の管腔濃度も有意に上昇した(図2F)。したがって、IEC内在性のHDAC3発現は、炎症性大腸菌CD4+ T細胞のプライミングを抑制するのに必須である。
図2
上皮性HDAC3欠損マウスのCD4+ T細胞は重篤な大腸炎を誘発する。(A)HDAC3FFおよびHDAC3ΔIECマウスから単離したナイーブCD4+ T細胞をRag1-/-宿主に移植したT細胞大腸炎モデルの実験概略図。(B-D)HDAC3FFマウスまたはHDAC3ΔIECマウスからT細胞を移入したRag1-/-宿主の体重(B)、結腸長(C)、およびH&E染色結腸切片(D)の変化。スケールバー:20μM。(F)リポカリン-2の糞便中濃度。(GおよびH)HDAC3FFマウスまたはHDAC3ΔIECマウスからT細胞を受け取ったRag1-/-宿主の大腸におけるcBir1+四量体特異的CD4+ T細胞の数。生細胞、CD45+、系統(CD11b-B220-Ly6G-、CD11c-CD8a-)、CD4+でゲーティング。(I)cBir1+CD4+T細胞のTh17(RORγt+)の頻度。(J-L)HDAC3FFマウスまたはHDAC3ΔIECマウスからT細胞を受け取った、水(CNV)または抗生物質(ABX)で処理したRag1-/-マウスの大腸における体重(J)、cBir1+四量体特異的CD4+ T細胞数(K)、およびRORγt+ cBir1+ CD4+T細胞の頻度(L)の変化。データは少なくとも2回の独立した実験の代表値であり、1群につき3-4匹のマウスを用いた。*P < 0.05、***P < 0.01、***P < 0.001、***P < 0.0001、無対2-tailed Studentのt検定(B-I)またはTukeyの多重比較検定付き1-way ANOVA(J-L)。
HDAC3ΔIECマウスから単離した大腸菌CD4+ T細胞をさらに調べるために、常在細菌叢発現抗原に対する特異性を評価した。HDAC3ΔIECマウスからCD4+ T細胞を受け取ったRag1-/-レシピエントは、HDAC3FF細胞を受け取った宿主と比較して、微生物叢特異的cBir1+CD4+ T細胞の上昇を示した(図2、GおよびH)。これらの実験から、移植後のcBir1+細胞の大部分はRORγt+ Th17細胞であることが示された(図2Iおよび補足図4B)。HDAC3ΔIECマウスにおけるこれらの細胞の偏った分化と同様に、HDAC3ΔIECマウスに由来する場合、HDAC3FF同腹仔コントロールと比較して、通性特異的T細胞の炎症性RORγt+ Th17細胞への分化頻度が高かった(図2I)。HDAC3ΔIECマウス由来の交感神経特異的T細胞は、FoxP3+ Tregへの分化が減少し、Th1細胞には差がなかった(補足図4B)。次に、これらの細胞が微生物叢に反応するかどうかを調べるため、広域抗生物質で常在菌を枯渇させることにより、T細胞移入試験を行った(50)。以前の結果と一致して、HDAC3ΔIECマウスからT細胞を受け取った微生物叢不全のレシピエントマウスでは、体重減少(図2J)の増加とcBir1+ Th17細胞の拡大が検出された(図2、KおよびL)。しかしながら、大腸炎による体重減少(図2J)およびcBir1+ Th17細胞は、微生物叢の枯渇とともに消失した(図2、KおよびL)ことから、このモデルでは、常在菌特異的Th17細胞が腸の炎症を促進していることが示された。これらのデータを総合すると、上皮性HDAC3の欠損は微生物特異的な結腸CD4+ T細胞の増加をもたらしたことから、上皮性HDAC3の発現が腸におけるCD4+ T細胞の発達の制御に必要であることが示された。
HDAC3は、IEC上のMHCIIの表面発現を制御している。サイトカインIFN-γは、CD4+ T細胞のTh1エフェクターへの分化を促進する一方で、Th17細胞を含む他のTh系への分化を抑制する(51, 52)。以前の研究で、上皮内HDAC3を欠損したマウスでは、細菌感染時に上皮内T細胞によるIFN-γ産生が減少することが示された(39)。しかし、定常状態ではIFN-γレベルは低く、有意差は検出されなかった(補図4、CおよびD)(39)。さらに、1型自然リンパ球(ILC1)はIFN-γを産生することができる。しかしながら、上皮性HDAC3の欠損は、ILC1sの頻度にも、大腸で優勢なILC系譜であるILC3sにも影響を与えなかった(補図4E)。さらに、外因性IFN-γを投与されたHDAC3ΔIECマウスでは、常在性特異的Th17細胞は増加したままであった(補足図4F)。したがって、IFN-γは、HDAC3ΔIECマウスで観察された通 常性特異的Th17細胞の基礎的差異の直接的または主要な原因とは考えにくい。
MHCIIを介した抗原提示は、抗原特異的CD4+ T細胞応答を指示するのに重要である。しかしながら、総MHCII+腸造血細胞(図3A)およびMHCIIhi腸樹状細胞やマクロファージを含む古典的抗原提示細胞の頻度は、上皮HDAC3欠失によって影響を受けなかった(図3、BおよびC)。しかし意外なことに、CD45+造血細胞ではなく、EpCAM+IECが腸-微生物叢界面でMHCIIの大部分を発現していることがわかった(図3、DおよびE)。IECは微生物の合図に反応するので、次に微生物叢がIEC内在性のMHCII発現を制御する役割を果たしているかどうかを評価した。他の研究(53-57)と一致して、CNVマウスから単離したIECはGFマウスと比較してMHCIIの表面発現が高かった(図3、FおよびG)。さらに、新生児GFマウスは、MHCIIのβ鎖をコードする遺伝子であるH2-Ab1の上皮発現が低いままであった(図3H)。しかしながら、上皮のH2-Ab1発現は、HDAC3の制御(図1C)と同様に、最初の微生物叢のコロニー形成後(図3H)に強固に誘導された。
図3
HDAC3はIEC上のMHCIIの発現を制御する。(A)結腸固有層における全MHCII+細胞の頻度。(BおよびC)HDAC3FFおよびHDAC3ΔIECマウスの大腸固有層における樹状細胞とマクロファージの頻度。生細胞、CD45+、MHCII+でゲーティング。(D)大腸内腔表面のMHCII+細胞。(E)Dにおける全MHCII+細胞の頻度。(FおよびG)GFおよびCNVマウスの大腸におけるMHCII+ EpCAM+細胞の頻度。(H)GFおよびCNV仔マウスの大腸から単離したIECにおけるH2-Ab1のmRNA発現。 IおよびJ)HDAC3FFおよびHDAC3ΔIECマウスの大腸におけるMHCII+ EpCAM+細胞の頻度。(KおよびL)HDAC3FFマウスおよびHDAC3ΔIECマウスの大腸から単離したIECにおけるH2-Ab1(K)およびCIITA(L)のmRNA。(M)タモキシフェン(4-OHT)で処理したHDAC3FFおよびHDAC3ΔIEC-INDオルガノイドにおけるHDAC3 mRNA。 N)IKK-16で処理したHDAC3FFおよびHDAC3ΔIEC-INDオルガノイドにおけるH2-Ab1 mRNA発現。データは少なくとも3回の独立した実験の代表値であり、1群につき3-5匹のマウスを用いた。*P < 0.05、***P < 0.01、***P < 0.001、*P < 0.0001は、対応のない両側スチューデントのt検定(E-M)またはTukeyの多重比較検定付き1元配置分散分析(N)により求めた。
興味深いことに、HDAC3の発現欠損は、大腸(図3、IおよびJ)および小腸(補足図5、AおよびB)における上皮表面MHCII発現を劇的に減少させた。HDAC3ΔIECマウスのIECはH2-Ab1遺伝子発現の減少を示し(図3Kおよび補足図5C)、MHCIIコード遺伝子の転写制御の変化を示唆した。HDAC3ΔIECマウスのIECでは、H2-Ab1遺伝子の近傍でヒストンアセチル化に有意な変化は見られなかったことから(補足図5D)、HDAC3はこの遺伝子を直接標的にしていないことが示唆された。クラスIIコアクチベーターCIITAの発現は、HDAC3ΔIECマウスのIECでは、欠損コントロールのIECに比べて低下していた(図3L)。CIITAはNF-κBによって制御されており、HDAC3は複数の細胞系においてNF-κBの活性化を促進することが示されている(58-61)。したがって、NF-κBがHDAC3依存性のMHCIIの制御を媒介するかどうかを調べるために、タモキシフェンがHDAC3の発現を有意に低下させるHDAC3FFおよび誘導性HDAC3ΔIEC-INDマウスモデルの大腸から腸オルガノイドを作製した(図3M)。NF-κB活性の阻害は、野生型オルガノイドにおけるH2-Ab1の発現を抑制した(図3N)。しかし、NF-κB阻害は、HDAC3を欠損したオルガノイドにおけるMHCII発現には影響しなかった(図3N)。これらのデータを総合すると、HDAC3は部分的にはNF-κBの活性化を通じて上皮MHCIIの発現を制御していることが裏付けられた。予想されたように、HDAC3FFオルガノイドはIFN-γ刺激後にH2-Ab1発現を上昇させたが(補足図5E)、HDAC3発現欠損オルガノイドでも同様の誘導が起こり(補足図5E)、HDAC3欠損上皮細胞ではIFN-γ依存的なMHCII制御がインタクトなままであることが支持された。IFN-γはin vivoで相乗的な役割を果たすかもしれないが、これらのオルガノイドのデータは、HDAC3が上皮MHCII発現を制御する上皮内在性のNF-κB依存的なメカニズムを示唆している。
上皮性MHCIIは、常在菌特異的CD4+ T細胞と腸の炎症を制限する。上皮性MHCIIは、防御的免疫応答と有害免疫応答の両方において機能することが示唆されているが(53, 57, 62, 63)、腸内常在菌特異的T細胞応答と微生物が誘発する炎症の制御におけるその役割は不明なままであった。そこで、このことを検証するために、H2-Ab1を欠損させたマウス(MHCIIFF)とビリンプロモーターの下流でCreリコンビナーゼを発現させたマウスを交配させ、IECにおけるMHCIIを条件付きで欠損させたマウス(MHCIIΔIEC)を作製した(64, 65)。MHCIIΔIECマウスのIECにおけるMHCII発現の有意な低下は、mRNA(補足図6A)および表面タンパク質分析(補足図6B)によって確認された。MHCII発現の消失はIECに限定され、CD45+細胞でのレベルは同程度であった(補足図6C)。興味深いことに、古典的抗原提示細胞におけるMHCIIの活性化作用とは対照的に、IEC内在性MHCIIの欠損は、結腸(図4、AおよびB)および小腸(補足図6D)における常在菌特異的cBir1+CD4+T細胞の蓄積を上昇させることがわかった。MHCIIΔIECマウスの腸では、常在菌特異的Tregが減少し(図4C)、常在菌特異的Th17細胞が増加した(図4D)が、微生物特異的Th1細胞とTfh細胞は同程度であった(補足図6E)。重要なことに、腸内細菌叢の組成と多様性は、cBir1遺伝子を保有する細菌の割合を含めて、MHCIIΔIECマウスと欠損同腹仔コントロールで同様であった(補足図7、A-C)(補足図7、DおよびE)。したがって、MHCIIΔIECマウスの腸内におけるcBir1常在菌特異的T細胞の増加は、常在菌の組成の変化を反映していない。
図4
上皮MHCIIは常在菌特異的T細胞を制御する。(AおよびB)MHCIIFFマウスおよびMHCIIΔIECマウスの大腸におけるcBir1+特異的CD4+ T細胞数。(CおよびD)MHCIIFFおよびMHCIIΔIECマウスの大腸におけるFoxP3+(C)および RORγt+(D)cBir1+CD4+T細胞の頻度。(E)2W1S-カンジダ・アルビカンス常在菌コロニー形成の図。(F)MHCIIFFおよびMHCIIΔIECマウスの大腸におけるC. albicans-2W1S+特異的CD4+細胞数。(G)2W1Sペプチド摂食モデルの図。(H)MHCIIFFおよびMHCIIΔIECマウスの大腸における2W1S+特異的CD4+細胞数。(I)MHCIIFFおよびMHCIIΔIECマウスにおける直腸脱の頻度。(J)MHCIIFFマウスおよび脱腸したMHCIIΔIECマウス(MHCIIΔIEC)のH&E染色大腸切片。スケールバー:20μM。(KおよびL)MHCIIFFおよびMHCIIΔIECの便における骨髄系細胞浸潤の頻度(K)およびリポカリン-2レベル(L)。(M-O) コントロールマウスおよび脱出したMHCIIΔIECマウスとHDAC3ΔIECマウスの大腸におけるcBir1+特異的CD4+ T細胞数(M)とFoxP3+(N)およびRORγt+(O)cBir1特異的T細胞の頻度。テトラマー細胞は、生細胞、CD45+、系統(CD11b-B220-Ly6G-、CD11c-CD8a-)、CD4+でゲートされる。データは、少なくとも2回の独立した実験(A-H)の代表値、または少なくとも2回の独立した実験(I-O)からプールしたもので、1群あたり3-6匹のマウス。P < 0.05、***P < 0.01、**P < 0.001は、対応のない両側Studentのt検定(B-HおよびL-O)またはMantel-Cox検定(I)。
この常在菌特異的反応が別の微生物抗原でも起こるかどうかを調べるために、マウスはI-Eαエピトープの2W1S55-68変異体を発現するCandida albicansでもコロニー形成された(図4E)(16, 17)。cBir1+CD4+T細胞の制御と一致して、MHCIIΔIECマウスはMHCIIFFマウスと比較して、C. albicans-2W1S特異的CD4+T細胞数の増加を示した(図4F)。さらに、2W1Sペプチド経口投与後の2W1S+CD4+T細胞の腸内蓄積を制限するためには、上皮MHCIIが必要であった(図4、GおよびH)(66)。上皮性MHCIIが組織内在性T細胞を制御するメカニズムを調べるために、微生物特異的T細胞の増殖、生存、アネルギーをMHCIIFFマウスとMHCIIΔIECマウスで比較した。上皮性MHCIIの欠損は、増殖マーカーKi67(補足図8A)とアネルギーマーカーFR4およびCD73(補足図8、BおよびC)に最小限の差異をもたらした。驚くべきことに、MHCIIΔIECマウス由来のcBir1+CD4+T細胞は、アポトーシスマーカーBim(補足図8、DおよびE)およびアネキシンV(補足図8F)の減少を示し、上皮性MHCIIがアポトーシスを促進し、MHCIIΔIECマウスにおけるそれらの局所的蓄積につながる可能性を示した。
IEC内在性HDAC3欠損マウスでは、慢性腸炎に対する感受性が上昇した(補足図1)。驚くべきことに、HDAC3ΔIECマウスと同様に、上皮性MHCII欠損マウスもまた、腸の炎症を示す直腸脱の発生率が加齢とともに増加した(図4I)。さらに、脱腸したMHCIIΔIECマウス(MHCIIΔIEC)は、慢性大腸炎に一致する病態の増加(図4J)、骨髄系細胞の浸潤の増加(図4K)、および糞便リポカリン-2の上昇(図4L)を示した。興味深いことに、脱腸傾向が増加したMHCIIΔIECマウスおよびHDAC3ΔIECマウスは、微生物特異的FoxP3+トレグ(図4N)の減少および常在菌特異的RORγt+炎症性Th17細胞(図4O)の増加を反映した、常在菌特異的CD4+ T細胞(図4M)のレベル上昇によって特徴付けられた。これらのデータを総合すると、上皮HDAC3は、微生物叢によって誘導されるMHCII指向性常在菌特異的免疫と炎症を制御する上で重要である可能性が示唆される。
HDAC3は、微生物叢による上皮依存性常在菌特異的免疫の制御を可能にする。MHCII依存性の腸管炎症と常在菌特異的免疫における微生物叢の必要性を検証するために、MHCIIΔIECマウスとコントロールのMHCIIFFマウスの飲料水に、常在菌を著しく減少させる広域抗生物質を補充した(50)。先の結果(図4I)と一致して、MHCIIΔIECマウスは直腸脱の有病率の増加を示したが(図5A)、微生物叢の枯渇はMHCIIΔIECマウスの自然腸炎を予防した(図5A)。さらに、微生物叢を欠乏させたMHCIIΔIECマウスは、常在菌特異的Th17細胞の蓄積(図5B)とIL-17レベルの上昇(図5C)を示した。しかし、抗生物質で処理したMHCIIΔIECマウスは、MHCIIFFマウスと比較して、常在菌特異的Th17細胞の増加(図5B)やIL-17発現の増加(図5C)を示さなかった。これらのデータを総合すると、上皮のMHCIIに依存した腸の炎症と常在菌特異的免疫応答の引き金となる微生物叢の必要性が浮き彫りになった。
図5
HDAC3は微生物叢が上皮依存的な常在菌特異的免疫を制御することを可能にする。(A)コントロールマウス(CNV)と抗生物質投与マウス(ABX)のMHCIIFFおよびMHCIIΔIECマウスにおける直腸脱の頻度。(BおよびC)対照およびABX処理MHCIIFFおよびMHCIIΔIECマウスの大腸におけるRORγt+ cBir1+特異的CD4+ T細胞の頻度(B)およびIL-17 mRNA発現(C)。(DおよびE)GF-およびCNV-HDAC3FFおよびHDAC3ΔIECマウスの大腸におけるMHCII+ EpCAM+細胞の頻度。(F-H)GF-およびCNV-HDAC3FFおよびHDAC3ΔIECマウスから単離したcBir1+四量体特異的CD4+ T細胞(F)の数、およびFoxP3+(G)およびRORγt+(H)cBir1+ T細胞の頻度。cBir1+四量体細胞は、生きたCD45+、系統(CD11b-B220-Ly6G-、CD11c-CD8a-)、CD4+でゲートされる。データは少なくとも2回の独立した実験の代表値で、1群につき3-5匹のマウスを使用。*P<0.05、***P<0.01、***P<0.001、**P<0.0001、Mantel-Cox(A)またはTukeyの多重比較検定付き1元配置分散分析(B-H)。
微生物叢による上皮細胞および免疫細胞のこの制御を媒介するのにHDAC3が必要かどうかを調べるために、GF由来のHDAC3FFおよびHDAC3ΔIECマウスをCNV-HDAC3FFおよび-HDAC3ΔIECマウスと比較した。野生型CNVマウスおよびGFマウスを用いた以前のデータ(図3、FおよびG)と一致して、IEC内在性MHCII発現は、GF-HDAC3FFマウスと比較して、CNV-HDAC3FFコントロールで有意に誘導されたが(図5、DおよびE)、CNV-HDAC3ΔIECマウスはMHCIIをアップレギュレートできなかった(図5、DおよびE)。GFマウスでは、上皮性HDAC3発現の喪失はIEC上のMHCIIのタンパク質レベルに影響を与えなかった(図5、DおよびE)ことから、微生物叢依存的な上皮性MHCIIの制御を媒介するHDAC3の特異的必要性が示された。さらに、CNV-HDAC3ΔIECマウスは、CNV-HDAC3FF同腹仔コントロールと比較して、cBir1+通 常菌特異的FoxP3+の減少および通 常菌特異的Th17細胞の上昇を示した(図5、F-H)。しかしながら、GF-HDAC3ΔIECは、GF-HDAC3FFマウスと同程度の常在特異的TregおよびTh17細胞レベルを有していた(図5、F-H)。これらのデータは、微生物叢が常在菌特異的免疫を制御するためにHDAC3が必要であることを示している。これらの知見を総合すると、微生物叢のコロニー形成は、局所組織環境において直接常在菌特異的免疫応答を減弱させる上皮HDAC3/MHCII制御経路を介して、常在菌の自己寛容を誘導できることが明らかになった(補足図9)。
考察
微生物叢は宿主免疫系の発達と教育に不可欠である。しかしながら、微生物叢に対する不適切な免疫反応は、いくつかの慢性炎症疾患の根底にあり、微生物叢特異的免疫がどのように制御されているかを理解する必要性を強調している。本研究では、微生物叢の初期コロニー形成が、上皮性HDAC3発現のアップレギュレーションと常在菌特異的T細胞の拡大をもたらすことを見いだした。IECにおけるHDAC3の選択的欠失は、上皮性MHCII発現の低下と、上皮性MHCIIによって制御されている常在菌特異的CD4+T細胞の制御に障害をもたらした。重要なことに、HDAC3またはMHCIIの上皮発現の消失は、通性特異的Th17細胞の増加と同時に、通性特異的Tregの減少をもたらした。これらの結果は、常在菌特異的T細胞が健常人の寛容細胞から活動性クローン病患者の炎症性IL-17分泌細胞へと切り替わることを示唆する最近のヒトのデータと一致している(14, 67)。HDAC3ΔIECマウスモデルとMHCIIΔIECマウスモデルでは、常在菌の組成は異なるが、両モデルともcBir1+常在菌特異的CD4+ T細胞の増加は同様であった。微生物特異的T細胞の宿主内在性制御と一致して、HDAC3は微生物特異的T細胞の上皮MHCII依存性制御を微生物叢が誘導するのに必要であった。したがって、常在微生物は、常在菌特異的CD4+ T細胞サブセットの局所動態と微生物叢感受性の疾患に対する感受性を制御するHDAC3依存性の上皮性MHCII経路を誘導することによって、自己寛容を指示することができる(補足図9)。
微生物特異的CD4+ T細胞は、胸腺(68)と末梢粘膜組織(15, 69-71)で中心的に生成される。自己反応性CD4+ T細胞は胸腺のネガティブセレクションによって制限されることがよく知られているが(72-74)、常在菌特異的細胞を制御するメカニズムは、あまりよく理解されていない。以前の研究では、活性化した常在菌特異的T細胞のネガティブセレクションは、MHCIIを発現するグループ3の自然リンパ球(ILC3)によって媒介されることが報告されている(75, 76)。ILC3は腸管内腔表面では比較的まれな細胞であるが、我々はIECが腸管MHCIIの主要な供給源であることを示している。上皮細胞に特異的なHDAC3依存性MHCII発現は、常在菌特異的Th17の蓄積を制限し、したがって、常在菌特異的CD4+ T細胞を制御し、微生物叢に対する病原性応答を減衰させるための、管腔表面における支配的なメカニズムである可能性がある。常在菌特異的T細胞の蓄積を制御することに加え、上皮のHDAC3またはMHCIIの欠損は、RORγt+抗原提示細胞を用いた最近の観察結果(77-79)と同様に、常在菌特異的Tregの有意な減少をもたらした。しかしながら、微生物叢に対するCD4+ T細胞の応答は、HDAC3に限定されない生体内の複数のシグナル伝達経路を反映している可能性が高い。このように、異なる非古典的細胞における抗原提示経路の相補的かつ本質的な役割は、健康な腸内組織の恒常性を確立し、維持するために極めて重要であると思われる。
腸の健康と疾病の制御における上皮性MHCIIに関する研究は、様々な、あるいは相反する結果をもたらしており、この制御の文脈依存的な役割を浮き彫りにしている。これまでの研究では、上皮性MHCIIを欠損させると、T細胞による腸炎や移植片対宿主病のモデルマウスが保護されることが示唆されている(53, 62)。対照的に、上皮性MHCIIはCD4+ T細胞によるTregの大量発生とIL-10の発現を促進し(57, 80)、CD4+ T細胞の活性化を制限する(81)という報告もある。さらに、IECは限られたコスティミュレイトリー分子を発現しており(82-84)、上皮性MHCIIの発現は、古典的な抗原提示細胞とは異なり、寛容原性T細胞応答を促進する可能性が示唆されている。我々のデータは、上皮性MHCIIが常在菌特異的T細胞応答を制御することによって腸の恒常性を促進するという、後者の研究による予測と実際に一致している。実際、上皮のHDAC3依存性MHCIIが欠損すると、腸内常在菌特異的T細胞が減少し、同時にTh17細胞が増加する。Tuganbaevら(57)の予測と一致して、我々の2W1Sのデータもまた、上皮性MHCIIが摂取抗原に向けられた腸管免疫応答を制御している可能性を支持している。このように、上皮性MHCII発現細胞は、常在微生物と食物抗原の両方に対する免疫寛容をより広範に制御し、腸の病的炎症を制御する可能性がある。
HDAC3依存性の上皮性MHCIIが欠損すると、常在菌特異的Th17活性が上昇し、微生物叢主導性の腸内炎症に対する感受性が上昇した(補足図9)。IFN-γはTh1応答を増幅する一方で、Th17細胞を含む他のThサブセットの分化と機能を阻害する(51, 52)。さらに、IL-17はTh1の分化を制限しながら、IFN-γの産生を抑制することもできる(85)。しかし、我々の系では、IFN-γはHDAC3ΔIECマウスで観察されたTh17表現型を駆動する主要因ではない。さらに、上皮性MHCIIを欠損したマウスでは、常在菌特異的Th17細胞の増加も認められた。驚くべきことに、活動性のIBD患者では、健常対照群と比べて、機能的に異なる微生物反応性CD4+ T細胞が存在し、IL-17を高濃度に分泌する(14, 26, 28-30, 67)。IL-17は、IBDを含むいくつかの自己免疫疾患や炎症性疾患の発症や増悪に関係している(28, 30, 86)。さらに、IL-17シグナル伝達が阻害されたマウスモデルは腸の炎症から保護されており(87, 88)、腸の炎症におけるIL-17の役割を裏付けている。さらに、微生物特異的Th17細胞はT細胞依存性大腸炎を引き起こすのに必要であり(89)、RORγt-またはIL-17欠損T細胞の移入は腸炎症の発症を予防する(90)。特異的四量体陽性細胞の希少性から、内因性常在菌特異的T細胞を直接移入することは不可能であるが、cBir1+ Th17細胞の増殖には微生物叢の存在が必要であったことから、常在菌反応性Th17が腸の炎症を誘導するのに重要であることが示唆された。しかし、IBD患者のIL-17を減少させる臨床試験は効果がなく、場合によっては病状を悪化させた(91)。腸のホメオスタシスを制御するIL-17のこのような多面的な役割から、IL-17そのものを広く中和するのではなく、特定のIL-17産生因子を標的とすることが、より効果的な治療法である可能性が示唆される。実際、他の細胞型からのIL-17産生を維持しつつ、常在菌特異的Th17細胞を標的とする治療薬が腸の炎症を抑制することが、これまでの研究で示されている(92)。我々のデータは、HDAC3活性の亢進によって上皮のMHCII発現を促進することで、炎症性常在菌特異的T細胞をさらに制限し、健康な腸の恒常性を促進する可能性を示唆している。
微生物叢と哺乳類免疫細胞間のクロストークには、パターン認識受容体の関与と微生物叢由来の代謝産物シグナル伝達を介したコミュニケーションが関与している。我々のデータは、微生物叢に感受性のある酵素HDAC3が腸の上皮性MHCII発現を制御するという、上皮制御の明確なレベルを明らかにしている。実際、上皮性HDAC3の欠損は、腸における微生物叢依存的なMHCII発現を消失させるのに十分であった。これまでの研究で、IEC内在性のMHCII発現には微生物叢が必要であることが示されている(53-57)。これらの研究の多くは、免疫細胞がIFN-γシグナル伝達を介してどのように上皮性MHCIIを誘導するかに焦点を当てている(53, 93, 94)。試験管内での研究に用いられたIFN-γの量は、腸内の基礎的な恒常性濃度に比べて比較的高い。しかし、この限界にもかかわらず、我々の研究は、微生物叢がHDAC3を介して上皮MHCIIの発現を促進する、上皮細胞内在性の新たなメカニズムを示唆している。加えて、正統的パターン認識経路の破綻は、腸管バリアーの完全性の喪失と炎症の亢進を引き起こし(95-97)、TLRシグナルアダプターであるMyD88とTRAFは、特に小腸において上皮性MHCIIの発現を誘導することが示されている(53)。MyD88/TRAFシグナルはNF-κBの活性化を促進し、CIITAとMHCIIを駆動することができるが、HDAC3はNF-κBの活性化を制御することが分かっている(58, 59, 61, 98, 99)。我々のデータは、NF-κBがHDAC3依存的に上皮性MHCIIの発現を誘導することを示している。HDAC3は多面的な酵素であり、ヒストンや非ヒストンの標的を脱アセチル化して遺伝子発現や細胞機能を変化させることができる。従って、HDAC3ΔIECマウスで観察される転写の違いは、ヒストン、そして潜在的には非ヒストン標的や酵素非依存的役割の制御変化に起因する集団的な結果を表している(59, 60, 100-103)。このようなIEC内在性の機序にもかかわらず、上皮MHCIIを促進するIFN-γの相乗的役割、あるいはin vivoにおけるMHCIIのHDAC3依存的制御が、細胞内在性および細胞外経路の統合されたネットワークを反映していることを否定することはできない。
肺上皮細胞、皮膚ケラチノサイト、線維芽細胞など、いくつかの組織特異的な非造血細胞がMHCIIを発現し、組織内在性のCD4+ T細胞応答を制御するのに必要な機構を備えていることを示唆する証拠が増えつつある(104-106)。HDAC3のユビキタスな性質を考えると、HDAC3が他の非古典的抗原提示細胞においてもMHCII依存性の経路を促進することはもっともらしい。興味深いことに、HDAC3の組織特異的欠失は、糖尿病、心臓病、アルツハイマー病、IBDの慢性炎症性疾患モデルと関連している(37, 38, 107-109)。重要なことに、今回得られた知見は、造血系以外の上皮性MHCIIに対するHDAC3の誘導が、微生物叢によって大きく誘導され、局所的な自己免疫応答を減衰させるという、免疫制御に関する基本的な新しい考え方を明らかにした。これらの知見は、微生物叢が常在菌指向性免疫の指導に利用する宿主の中心的メカニズムを明らかにし、この制御経路が慢性炎症状態の治療の標的となり得ることを示唆している。
方法
マウス C57BL/6Jマウスはジャクソン研究所から購入し、シンシナティ小児病院メディカルセンター(Cincinnati Children's Hospital Medical Center:CCHMC)の特定病原体フリー従来型(CNV)施設で維持した。無菌(GF)マウスは、CCHMCのGnotobiotic Mouse Facilityのフレキシブルアイソレーターで維持し、オートクレーブ処理した飼料と水を与え、微生物がいないことをモニターした。HDAC3FF、HDAC3ΔIEC、およびHDAC3ΔIEC-INDマウスは、以前に記載されたように作製した(37)。H2-Ab1(MHCII)FFおよびRag1-/-マウスは、The Jackson Laboratoryから購入し、CCHMCで維持した。MHCIIFFマウスをC57BL/6J-Villin-Creと交配し、MHCIIΔIECマウスを作製した。性および年齢を一致させた同腹仔マウスをすべての研究に用いた。マウスは換気ケージシステムで1ケージあたり4匹まで飼育し、12時間明期/12時間暗期サイクルで、水と餌を自由に摂取できるようにした。すべてのマウス研究は、CCHMCのInstitutional Animal Care and Use Committeeの承認を得て実施された。これらのプロトコールは、米国公衆衛生局および農務省が制定した基準に従っている。すべての実験は、Animal Researchの定める基準に従った: In Vivo実験の報告(ARRIVE)。
マウス大腸炎および2W1Sモデル。T細胞移入大腸炎モデルについては、HDAC3FFマウスおよびHDAC3ΔIECマウスの脾臓および腸間膜リンパ節から、MojoSort Mouse CD4 Naive T Cell Isolation(BioLegend社)を介して、5×105個のナイーブCD4+ T細胞を単離した。細胞の純度をフローサイトメトリーで確認し、年齢と性別が一致したRag1-/-レシピエントにT細胞をi.p.注射した。抗生物質投与については、MHCIIFFおよびMHCIIΔIECマウスの仔マウスに、離乳時に1mg/mLのコリスチン(ミリポアシグマ社製)、1mg/mLのアンピシリン(ミリポアシグマ社製)、および5mg/mLのストレプトマイシン(ミリポアシグマ社製)を添加した水を与え、16週間抗生物質で維持した。Rag1-/-マウスは、T細胞移植の7-10日前から同じ抗生物質カクテルを投与され、その後、実験期間中抗生物質水で維持された。抗生物質水は7-10日ごとに交換した。PBSまたは10μgの組換えIFN-γ(PeproTech)をi.p.投与されたマウスは、24時間後に分析された。C. albicans-2W1Sのコロニー形成は、以前に記載されたように行った(16, 17, 110)。簡単に述べると、マウスはコロニー形成の2日前にアンピシリン-水(1 mg/mL)で前処理され、実験期間中はアンピシリン-水で維持された。マウスは、2W1S55-68ペプチドを発現する組換えC. albicansを口から滴下された(111)。マウスのコロニー形成は、コロニー形成の14日後に採取した糞便CFUのプレーティングによってモニターした。2W1Sの摂食については、MHCIIFFおよびMHCIIΔIECマウスに、0日目、2日目、および4日目に100μgの2W1Sペプチドを経口投与し、その後6日目に2W1S特異的細胞を前述のように採取した(66)。
細胞の分離。大腸を摘出、開腹し、PBSで洗浄した。IECについては、組織をあらかじめ温めたストリップバッファー(PBS、5%FBS、1mM EDTA、1mM DTT)に入れ、37℃、45°の角度で180rpmで15分間振盪しながらインキュベートした。薄層前膜の分離では、組織をPBSで洗浄してEDTAとDTTを除去し、あらかじめ温めた消化バッファー(1 mg/mL Collagenase/Dispase [MilliporeSigma]を加えたRPMI)中で37℃、45°の角度で180 rpmで30分間振盪しながらインキュベートした。インキュベーション後、組織をボルテックスし、70μmの細胞ストレーナーに通した。
フローサイトメトリー。FACSバッファー(2%FBS、0.01%アジ化ナトリウム、PBS)で希釈した以下の抗体を用いて、フローサイトメトリー用に細胞を染色した: BV711-抗CD326(EpCAM)(クローンG8.8、BioLegend)、BUV395-抗CD45.2(クローン104、BD Biosciences)、APC-またはFITC-抗MHCII(クローンM5/114.15. 2、eBioscience)、APC-eFluor 780-抗CD4(クローンRM4-5、eBioscience)、PE-Cy7-抗CD44(クローンIM7、eBioscience)、Alexa Fluor 647-またはBV650-抗RORγt(クローンQ31-378、BD Biosciences)、PerCP-eFluor 710-またはAPC-抗CD8a(クローン53-6. 7、eBioscience)、PerCP-Cy5.5-抗CD3(クローン17A2、eBioscience)、PerCP-eFluor 710-抗B220(クローンRA3-6B2、eBioscience)、PerCP-eFluor 710-抗Ly6G(クローン1A8-Ly6g、eBioscience)、PerCP-Cy5. 5-、eFluor 450-またはPE-抗CD11b(クローンM1/70、eBioscience)、PerCP-Cy5.5-抗CD11c(クローンN418、eBioscience)、BV650-抗CXCR5(クローン138D7、BioLegend)、Alexa Fluor 647-抗FR4(クローン12A5、BioLegend)、eFluor 450-抗CD73(クローンeBioTY/11. 8、eBioscience)、Alexa Fluor 488-抗Bim(C34C5、Cell Signaling Technology)、BV421-抗Tbet(クローン4B10、BioLegend)、FITC-抗CD90.2(クローン53-2. 1、eBioscience)、PE-Cy7-抗CD127(クローンA7R34、eBioscience)、eFluor 450-抗Ki67(クローンSolA15、eBioscience)、PE-抗IFN-γ(クローンXMG1.2、eBioscience)。サイトカイン染色では、細胞を50ngのPMAと1μgのイオノマイシンで37℃で3-4時間刺激した。細胞を、アネキシンV染色バッファー(BD Pharmingen)で希釈したAlexa Fluor 488またはAPC標識アネキシンV(eBioscience)で染色した。死細胞はFixable Aqua Dead Cell Stain Kit(Invitrogen)を用いて除いた。細胞内染色にはBD Fix/Permキットを用いた。クラスII制限四量体(cBir1: YSNANILSQ; および2W1S: EAWGALANWAVDSA)はPE結合で、NIH tetramer coreから提供された。テトラマー染色では、サンプルをテトラマー(1:100)およびFcブロック(抗マウスCD16/CD32、eBioscience)と共に室温で1時間インキュベートした。サンプルはBD LSRFortessaで取得し、FlowJo Software(Tree Star)で解析した。
RNAおよび定量PCR解析。RNAはRNeasy Kit(Qiagen)を用いて単離した。全組織からのRNAについては、サンプルをTRIzolでホモジナイズした。クロロホルムを加えて相分離し、イソプロパノールと混合してRNAを沈殿させた。cDNAはVerso逆転写酵素キット(Thermo Fisher Scientific)を用いて、製造元のプロトコールに従って合成した。SYBR Green(Applied Biosystems)を用いてリアルタイムPCRを行い、以下のマウスプライマー配列を用いて解析した: HPRTフォワード5′-GATTAGCGATGAACCAGGT-3′、HPRTリバース5′-CCTCCCATCTCCTTCATGACA-3′、H2-Ab1フォワード5′-TGTGAGTCCTGGTGACTGCCATTA-3′、 H2-Ab1リバース5′-TCGCCCATGAACTGGTACGAAA-3′、IL-17フォワード5′-ACCGCAATGAAGACCCTGAT-3′、 IL-17リバース5′-TCCCTCCGCATTGACACA-3′、HDAC3フォワード5′-TTGGTATCCTGGAGCTGCTT-3′、HDAC3リバース5′-GACCCGGTCAGTGAGGTAGAGA-3′、 CIITAフォワード5′-CCCTGCGTGTGATGGATGTC-3′、CIITAリバース5′-ATCTCAGACTGATCCTGGCAT-3′。
リポカリン-2ELISA。糞便ペレットを100mg/mLの濃度でPBS中でホモジナイズし、高速で10分間遠心分離した。上清を回収し、マウスLipocalin-2/NGAL ELISAキット(R&D Systems社製)を用いて、製造者の指示に従ってリポカリン-2濃度を測定した。
腸オルガノイド HDAC3FFマウスおよびHDAC3ΔIEC-INDマウスの大腸から、以前に記載されたように腸オルガノイドを作製した(38, 112)。手短に言えば、大腸を切開して小片にし、キレート化バッファー(PBS中2mM EDTA)中で、4℃で30分間、回転させながらインキュベートした。次に組織を振盪バッファー(PBS、43.3mMスクロース、54.9mMソルビトール)に移し、手で2-5分間振盪した。大腸陰窩を再懸濁し、オルガノイド培養培地(10mM HEPES、2mM l-グルタミン酸、40% L-WRN調整培地、1×N2サプリメント、1×B27サプリメント、50ng/mLマウスEGF、および10μM Y-27632 ROCK阻害剤を添加した60% Advanced DMEM/F12培地[Gibco, Thermo Fisher Scientific])とともにマトリゲル(Corning)中にプレーティングした。培養液は2-3日ごとにリフレッシュした。HDAC3欠失を誘導するために、オルガノイドを1μMのヒドロキシタモキシフェン(4-OHT、MilliporeSigma)で24時間処理した。オルガノイドをビヒクル(DMSO)または5μM IKK-16(Selleckchem)で24時間処理した。IFN-γ刺激では、オルガノイドを100 U/mLのマウス組み換えIFN-γ(BioLegend)で24時間処理した。インキュベーション後、オルガノイドをPBSで3回洗浄し、RNeasyキット(Qiagen)を用いて溶解した。
微生物叢解析。ショットガンメタゲノムシークエンシングのために、メーカーの推奨に従い、PowerFecal DNA isolation kit(Qiagen Inc.)を使用して約0.1gの便からDNAを抽出した。シーケンスライブラリーは、Nextera XTプロトコル(Illumina)を用いて微生物DNAから作成した。シーケンシングは、Illumina NovaSeq 6000マシンを用いて、サンプルあたり約4G塩基対の深さまで150bp DNAペアエンドリードを用いて行った。生配列データは、多重化を解除してFASTA形式に変換し、ダウンストリーム解析に供した。各サンプルのペアエンドシーケンスリードは、ヒトゲノムおよび約40,054の細菌、真菌、ウイルス、寄生虫ゲノムからなるカスタムゲノムデータベースに対してKraken (113)でアライメントされた。このデータベースは、ヒトゲノム(GR38Ch; https://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml)だけでなく、RefSeqゲノムデータベース(https://www.ncbi.nlm.nih.gov/refseq/)に登録されているすべての細菌、真菌、ウイルスから最初に作成された。NCBI AssembliesとPATRICのドラフトゲノムを含む追加ゲノムの追加には、手作業によるキュレーションが用いられた。グループ間の全体的なマイクロバイオーム組成の比較は、RのVeganパッケージ(114)を用いたmulti-response permutation procedure(MRPP)により行った。同腹のHDAC3FFマウスとHDAC3ΔIECマウス、およびMHCIIFFマウスとMHCIIΔIECマウスから得られたマイクロバイオームショットガンシーケンスリードは、プログラムDiamond(116)のtblastコマンドとデフォルト設定を用いて、cBir1ペプチド配列(115)(AY551005.1)に対してアライメントした。この遺伝子の有病率は、マップされた100万個の細菌リードあたりのcBir1カウントとして表される。cBir1定量的PCR(qPCR)解析のために、QIAamp Fast DNA Stool Extract Kit(Qiagen)を用いて、製造元の説明書に従って便DNAを抽出した。細菌DNAは、以下のプライマーセットを用いたqPCR分析により、cBir1遺伝子の発現を評価した:cBir1フォワード5′-AAGTACTTTACGGCAGGCGG-3′、cBir1リバース5′-TCTGTTCCGTCAGCACCTAC-3′。
組織学的解析。大腸組織切片を4%パラホルムアルデヒドで4℃で一晩固定し、パラフィン包埋、切片化し、H&Eで染色した。組織学的スコアリングのために、スライドは以下のパラメーターに基づいて評価された:免疫細胞浸潤、1-5;粘膜肥厚/浮腫、1-5;陰窩長さ、1-5;陰窩脱落/浸食、1-5。
統計。結果は平均値±SEMで示した。2群間の検定には両側Studentのt検定を用いた。多重比較にはTukeyの検定による1-way ANOVAを用いた。脱出曲線はlog-rank Mantel-Cox検定を用いて評価した。結果はP < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001で有意とみなされた。統計的有意差の算出にはPrism version 7.0(GraphPad Software)を用いた。
研究の承認 すべての動物実験は、Cincinnati Children's Hospital Medical CenterのInstitutional Animal Care and Use Committeeにより承認された。
著者貢献
EME、SSW、TAが本試験のコンセプトを立案し、試験方法を設計した。EME、TYA、VW、TR、BJD、LE、JW、DBHが実験を行った。EME、TYS、TR、DBHがデータを解析。EME、TA、SSWが原稿の執筆、査読、編集を行った。
補足資料
補足データを見る
謝辞
NIH Tetramer Core Facility(NIH-NIAID契約75N93020D00005の支援)には、MHCモノマーおよびテトラマー試薬を提供していただいた。Way、Qualls、Deshmukhの各研究室には有益な議論を、Allenghat研究室のメンバーには原稿の批判的な読解をいただいた。CCHMC Veterinary Services、Research Flow Cytometry Core、Confocal Imaging Core、Pathology Research Coreのサービスおよび技術協力に感謝する。本研究はNIH(TAにDK114123、DK116868、SSWにDP1AI131080、EMEにF32AI147591)、およびTAにKenneth Rainin財団の助成を受けた。SSWとTAはそれぞれBurroughs Wellcome FundからInvestigators in the Pathogenesis of Infectious Disease Awardを授与されている。このプロジェクトは、Public Health Serviceの助成金P30DK078392の一部を受けた。
連絡先 Theresa Alenghat, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, MLC 7038, Cincinnati, Ohio 45229, USA. 電話: 513.803.7498; Email: Theresa.Alenghat@cchmc.org. または下記まで: Sing Sing Way, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, MLC 7017, Cincinnati, Ohio 45229, USA. 電話 513.636.7603; Email: SingSing.Way@cchmc.org.
脚注
利益相反: 著者らは利益相反が存在しないことを宣言している。
Copyright: © 2023, Eshleman et al. これは、Creative Commons Attribution 4.0 International Licenseの条件の下で公開されたオープンアクセス論文である。
参考情報 J Clin Invest. 2023;133(4):e162190. https://doi.org/10.1172/JCI162190.
参考文献
Hill DA, Artis D. 腸内細菌と免疫細胞のホメオスタシス制御。Annu Rev Immunol. 2010;28(1):623-667.
この記事を見る CrossRef PubMed Google Scholar
ベルカイド Y, ハリソン OJ. 恒常性免疫と微生物叢。Immunity. 2017;46(4):562-576.
この記事を見る CrossRef PubMed Google Scholar
本田和彦、Littman DR. 適応免疫恒常性と疾患における微生物叢。Nature. 2016;535(7610):75–84.
この記事を見る CrossRef PubMed Google Scholar
腸内における微生物叢、上皮、免疫細胞の相互作用。Annu Rev Immunol. 2020;38:23-48.
この記事を見る CrossRef PubMed Google Scholar
親しみやすい真菌:常在菌カンジダ・アルビカンスとの共生。Trends Immunol. 2022;43(9):706-717.
この記事を見る CrossRef PubMed Google Scholar
乳幼児の腸内細菌叢における微生物コンソーシアムの継代。この論文では、腸内細菌叢の形成過程と、その形成に必要な微生物群集を明らかにした。
この記事を見る PubMed Google Scholar
Yatsunenko T, et al. ヒト腸内細菌叢の年齢と地理的変化。Nature. 2012;486(7402):222–227.
この記事を見る クロスリーフ PubMed Google Scholar
健康と病気における微生物叢と免疫の相互作用。細胞研究(Cell Res)2020;30(6):492-506。
この論文を見る CrossRef PubMed Google Scholar
生後1年間のヒト腸内細菌叢の動態と安定化。細胞宿主微生物。2015;17(5):690-703.
この記事を経由して表示します: CrossRef PubMed Google Scholar
本田和彦、リットマンDR. 感染症と炎症におけるマイクロバイオーム。Annu Rev Immunol. 2012;30(1):759-795.
この記事を見る CrossRef PubMed Google Scholar
ヒトにおける腸内細菌、バクテロイデス、ビフィズス菌、常在腸内細菌叢由来抗原に対するT細胞の特異性と交差反応性。Gut. 1999;44(6):812-818.
この記事を見る CrossRef PubMed Google Scholar
Duchmann R, et al. 常在腸内細菌叢に対する寛容は存在するが、活動性炎症性腸疾患(IBD)では壊れる。Clin Exp Immunol. 1995;102(3):448-455.
この記事を経由して表示します: PubMed Google Scholar
Ergin A, et al.クローン病および強直性脊椎炎患者における大腸菌蛋白に対する末梢Th1 CD4+T細胞応答の障害。J Clin Immunol. 2011;31(6):998-1009.
この記事を見る CrossRef PubMed Google Scholar
Hegazy AN, et al.腸内細菌叢に反応する血中および組織常在CD4+ t細胞は健常人に多く、炎症時には機能が変化する。Gastroenterology. 2017;153(153):1320–1337.
この記事を経由して見る PubMed Google Scholar
急性胃腸炎は長期にわたる微生物特異的T細胞応答を誘導する。Science. 2012;337(6101):1553–1556.
この記事を見る クロスリーフ PubMed Google Scholar
カンジダ・アルビカンス(Candida albicans)は全身性のTh17免疫応答を制御する。Cell Host Microbe. 2019;25(3):404-417.
この論文を見る CrossRef PubMed Google Scholar
Jiang TT, et al. 宿主真菌は腸内細菌の保護効果を再現する。Cell Host Microbe. 2017;22(6):809-816.
この記事を見る CrossRef PubMed Google Scholar
Su LF, et al.ウイルス特異的CD4(+)メモリーフェノタイプT細胞は未曝露成人で豊富である。Immunity. 2013;38(2):373-383.
この記事を見る クロスリーフ PubMed Google Scholar
未曝露の献血者におけるHIV特異的ナイーブおよびメモリーCD4(+)T細胞のプロテオームワイド解析。J Exp Med. 2014;211(7):1273–1280.
この記事を見る クロスリーフ PubMed Google Scholar
炎症性腸疾患。Annu Rev Immunol. 2010;28(12):573-621.
経由でこの記事を表示します: CrossRef PubMed Google Scholar
Tindemans I, et al. IBDにおけるT細胞介在性炎症の不均一性を解明する。Cells. 2020;9(1):110.
この記事を見る CrossRef PubMed Google Scholar
腸内細菌叢がヒトの健康に及ぼす影響:統合的見解。細胞。2012;148(6):1258–1270.
経由でこの記事を見る: CrossRef PubMed Google Scholar
常在細菌に対するエフェクターT細胞とメモリーT細胞の反応。Trends Immunol. 2013;34(6):299-306.
この記事を経由して表示します: クロスリーフ PubMed Google Scholar
炎症性腸疾患における腸の恒常性とその破綻。Nature. 2011;474(7351):298–306.
この記事を経由して表示します: CrossRef PubMed Google Scholar
自然発症大腸炎C3H/HeJBirマウスにおける腸内細菌抗原に反応するCD4+ T細胞:Tヘルパー細胞1型反応の増加と疾患移行能力。J Exp Med. 1998;187(6):855-864.
この記事を見る CrossRef PubMed Google Scholar
Annunziato F, et al. ヒトTh17細胞の表現型と機能的特徴。J Exp Med. 2007;204(8):1849–1861.
この記事を見る クロスリーフ PubMed Google Scholar
Alexander M, et al. ヒト腸内細菌の代謝はTh17の活性化と大腸炎を促進する。細胞宿主微生物。2022;30(1):17-30.
この記事を経由して表示します: CrossRef PubMed Googleスカラー
Gálvez J. ヒトIBDの病態におけるTh17細胞の役割。ISRN Inflamm. 2014;2014:1-14.
この記事を経由して表示します: PubMed Google Scholar
炎症性腸疾患におけるインターロイキン17の発現亢進。Gut. 2003;52(1):65-70.
この論文を見る CrossRef PubMed Google Scholar
炎症性腸疾患におけるTh17細胞:サイトカイン、可塑性、治療。J Immunol Res. 2021;2021:8816041.
この記事を見る PubMed Google Scholar
微生物叢由来シグナルの上皮感知。Genes Immun. 2021;22(5–6):237–246.
この論文を見る PubMedグーグルスカラー
ローDD. 警戒か破壊か?粘膜組織における恒常性M細胞と誘導性M細胞。Trends Immunol. 2017;39(3):185-195.
この記事を見る PubMed Google Scholar
Kulkarni DH, et al. Goblet cell associated antigen passages support the induction and maintenance of oral tolerance. Mucosal Immunol. 2020;13(2):271-282.
この論文を見る CrossRef PubMed Google Scholar
腸管M細胞:腸内細菌叢の疲れを知らないサンプラー。Traffic. 2020;21(1):34-44.
この記事を見る CrossRef PubMed Google Scholar
Tヘルパー細胞サイトカインは腸管幹細胞の再生と分化を調節する。Cell. 2018;175(5):1307–1320.
この記事を見る CrossRef PubMed Google Scholar
Kambayashi T, Laufer TM. 非定型MHCクラスII発現抗原提示細胞:樹状細胞に代わるものはあるか?Nat Rev Immunol. 2014;14(11):719-730.
この記事を見る CrossRef PubMed Google Scholar
ヒストン脱アセチル化酵素3は、常在細菌に依存した腸の恒常性を調整する。Nature. 2013;504(7478):153–157.
この記事を見る クロスリーフ PubMed Google Scholar
微生物由来の代謝産物が腸内のHDAC3活性を促進する。Nature. 2020;586(7827):108–112.
この記事を見る クロスリーフ PubMed Google Scholar
Navabi N, et al. 上皮性ヒストン脱アセチル化酵素3は、局所リンパ球活性化を調整することにより腸管免疫を指示する。Cell Rep. 2017;19(6):1165-1175.
この記事を見る クロスリーフ PubMed Google Scholar
腸内細菌叢はヒストン脱アセチル化酵素3を介して宿主の代謝の日内リズムをプログラムする。Science. 2019;365(6460):1428–1434.
経由でこの記事を見る: CrossRef PubMed Google Scholar
Aagaard K, et al. Una destinatio, viae diversae: Is exposure to the vaginal microbiota confer health benefits to the infant, and does lack of exposure confer disease risk? EMBO Rep.
この記事を見る CrossRef PubMed Google Scholar
ラッセルSLら:早期の抗生物質による微生物叢の変化はアレルギー性喘息への感受性を高める。EMBO Rep.
この論文を見る クロスリーフ PubMed Google Scholar
幼少期の微生物叢によるコロニー形成が免疫系を形成する。Science. 2016;352(6285):539–544.
この記事を見る CrossRef PubMed Google Scholar
微生物叢に対する離乳期の反応は、成人における免疫病理に対する抵抗性に必要である。Immunity. 2019;50(5):1276–1288.
経由でこの記事を見る: CrossRef PubMed Google Scholar
Wang T, et al. 幼少期の抗生物質曝露と宿主の健康: 微生物-免疫相互作用の役割。Semin Perinatol. 2020;44(8):151323.
この記事を経由して表示します: CrossRef PubMed Google Scholar
Gray J, et al. 腸内常在菌は肺粘膜免疫を媒介し、新生マウスの感染抵抗性を促進する。Sci Transl Med. 2017;9(376):1-14.
この記事を見る PubMed Google Scholar
Ostanin DV, et al.慢性大腸炎のT細胞移入モデル:概念、考察、および取引のコツ。Am J Physiol Gastrointest Liver Physiol.
この記事を見る PubMedのGoogleのスカラー
免疫不全マウスにおける制御性T細胞の枯渇による炎症性腸疾患の誘発。Curr Protoc Immunol. 1999;30(1):1-10.
この記事を見る PubMed Google Scholar
Powrie BF, et al. CD45RB高値CD4+T細胞とCD45RB低値CD4+T細胞間の制御性相互作用は、防御的細胞媒介免疫と病原性細胞媒介免疫のバランスに重要である。J Exp Med. 1994;179(2):589-600.
この記事を見る CrossRef PubMed Google Scholar
Vétizou M, et al. CTLA-4遮断による抗癌免疫療法は腸内細菌叢に依存する。Science. 2015;350(6264):1079–1084.
経由でこの記事を見る: CrossRef PubMed Google Scholar
インターロイキン17を産生することにより組織炎症を制御するCD4 T細胞の別系統。Nat Immunol. 2005;6(11):1133–1141.
この記事を経由して表示します: CrossRef PubMed Google Scholar
インターロイキン17を産生するCD4+エフェクターT細胞は、Tヘルパー1型や2型とは異なる系統を経て発達する。Nat Immunol. 2005;6(11):1123–1132.
この記事を見る CrossRef PubMed Google Scholar
腸上皮によるMHCクラスII抗原提示は移植片対宿主病を発症させ、微生物叢の影響を受ける。Immunity. 2019;51(5):885-898.
この記事を経由して見る CrossRef PubMed Google Scholar
γδTCRを有する上皮内リンパ球はマウス小腸上皮におけるクラスII主要組織適合複合体分子の発現を制御する。Epithelial Cell Biol.
この記事を見る PubMed Google Scholar
分節性糸状菌は上皮内リンパ球を活性化し、MHCクラスII分子とフコシルアジアロGM1糖脂質を誘導する。Microbiol Immunol. 1995;39(8):555-562.
この記事を経由して表示します: CrossRef PubMed Google Scholar
マウス腸管上皮細胞の細胞増殖と分化における上皮内リンパ球の生理的役割。免疫学。1999;97(1):18-25.
この記事を経由して表示します: CrossRef PubMed Google Scholar
Tuganbaev T, et al. 食事は小腸の微生物-上皮-免疫恒常性と腸炎を日周的に制御する。Cell. 2020;182(6):1441–1459.
この記事を見る CrossRef PubMed Google Scholar
IL-1シグナル伝達におけるヒストン脱アセチル化酵素3のコアクチベーターとしての役割は、p65 NF-κBの脱アセチル化に関与する。Nucleic Acids Res.
この記事を見る クロスリファレンス PubMed Google Scholar
マクロファージにおける炎症性遺伝子発現プログラムに対するヒストン脱アセチル化酵素Hdac3の必要性。Proc Natl Acad Sci U S A. 2012;109(42):2865-E2874.
この記事を見る PubMed Google Scholar
Nguyen HCB, et al. HDAC3活性の二律背反的関与が炎症反応を支配する。Nature. 2020;584(7820):286–290.
この論文を見る CrossRef PubMed Google Scholar
HDAC3選択的阻害剤RGFP966は、NF-κB p65転写活性を減弱させることにより、RAW264.7マクロファージおよびマウス精密切断肺スライスにおいて抗炎症作用を示す。Biochem Pharmacol. 2016;108:58-74.
View this article via: CrossRef PubMed Google Scholar
MHCIIの腸管上皮発現が化学物質、T細胞誘導性、感染性大腸炎の重症度を決定する。Gastroenterology. 2020;159(4):1342–1356.
この記事を見る CrossRef PubMed Google Scholar
インターフェロンγは腸上皮細胞にMHCクラスIIの発現を誘導し、マウスを大腸炎から保護する。PLoS One. 2014;9(1):1-10.
この記事を経由して表示します: PubMed Google Scholar
主要組織適合性IA-β鎖遺伝子の条件付きヌル対立遺伝子。Genesis. 2002;32(2):152-153.
この記事を見る クロスリーフ PubMed Google Scholar
El Marjou F, et al. 腸上皮における組織特異的かつ誘導可能なCre媒介組換え。Genesis. 2004;39(3):186-193.
この記事を見る クロスリファレンス PubMed Google Scholar
食物の免疫寛容はCD4+ T細胞の機能不全の層によって媒介される。Nature. 2022;607(7920):762–768.
この論文を見る CrossRef PubMed Google Scholar
クローン病では、常在菌由来のエピトープに対するCD4+ T細胞応答が寛容状態から炎症状態へと移行する。Immunity. 2022;55:1-15.
この記事を見る CrossRef PubMed Google Scholar
Zegarra-Ruiz DF, et al. 腸内細菌特異的T細胞の胸腺での発生。Nature. 2021;594(7863):413–417.
この論文を見る クロスリーフ PubMed Google Scholar
ベルカイドY、ハンドTW. 免疫と炎症における微生物叢の役割。Cell. 2014;157(1):121-141.
この記事を見る クロスリーフ PubMed Google Scholar
Xu M, et al. c-MAF依存性制御性T細胞は腸内病原体に対する免疫寛容を媒介する。Nature. 2018;554(7692):373–377.
経由でこの記事を見る: CrossRef PubMed Google Scholar
Yang Y, et al. 常在細菌抗原に対する腸管TH17細胞の集束特異性。Nature. 2014;510(7503):152–156.
この論文を見る クロスリーフ PubMed Google Scholar
T細胞レセプターの発達中のT細胞のポジティブ選択とネガティブ選択における役割。Science. 1990;248(15):1335–1341.
この論文を見る PubMed Google Scholar
Mathis D, Benoist C. Aire. Annu Rev Immunol。2009;27:287-312.
経由でこの記事を表示します: クロスレフ PubMed Google Scholar
T細胞レパートリーの正選択と負選択:胸腺細胞は何を見ているのか(そして見ていないのか)。Nat Rev Immunol. 2014;14(6):377-391.
この記事を見る クロスリーフ PubMed Google Scholar
自然リンパ球は腸内常在菌に対するCD4+ T細胞応答を制御する。Nature. 2013;498(7452):113–117.
この記事を見る CrossRef PubMed Google Scholar
免疫寛容。グループ3自然リンパ球は、常在細菌特異的CD4+ T細胞の腸管選択を媒介する。Science. 2015;348(6238):1031–1035.
経由でこの記事を見る: CrossRef PubMed Google Scholar
ILC3s select microbiota-specific regulatory T cells to establish tolerance in the gut. Nature. 2022;610(7933):744–751.
この記事を見る クロスリーフ PubMed Google Scholar
新規抗原提示細胞は腸内細菌叢に対するTreg依存的寛容を与える。Nature. 2022;610(7933):752–760.
この論文を見る クロスリーフ PubMed Google Scholar
RORγt+細胞が腸内細菌叢特異的Treg細胞の分化を誘導する。Nature. 2022;610(7933):737–743.
この論文を見る CrossRef PubMed Google Scholar
Westendorf AM, et al. CD4+Foxp3+制御性T細胞の膨張は、局所樹状細胞とは無関係に、腸上皮細胞との抗原主導的相互作用によって誘導される。Gut. 2009;58(2):211-219.
この記事を見る CrossRef PubMed Google Scholar
Cruickshank SM, et al. 大腸上皮細胞を介したCD4 T細胞活性化の抑制。Gut. 2004;53(5):678-684.
この論文を見る CrossRef PubMed Google Scholar
Byrne B, et al. ヒト十二指腸上皮細胞はコスティミュレイトリー分子ではなく抗原提示分子成分を構成的に発現している。Hum Immunol. 2002;63(11):977-986.
この記事を見る CrossRef PubMed Google Scholar
なぜ腸上皮細胞はMHCクラスIIを発現するのか?免疫学 2021;162(4):357-367.
この論文を見る クロスリーフ PubMed Google Scholar
腸管細胞とリンパ球におけるB7 mRNAの制御の違い。免疫学。1993;79(3):434-438.
この論文を見る PubMed Google Scholar
T細胞介在性腸炎症におけるインターロイキン17Aの保護機能。Nat Immunol. 2009;10(6):603-609.
この記事を見る CrossRef PubMed Google Scholar
炎症におけるTヘルパー17細胞エフェクターサイトカインの生物学的機能。Immunity. 2008;28(4):454-467.
この記事を見る クロスリーフ PubMed Google Scholar
実験的大腸炎におけるTH17細胞の誘導とIL-17AおよびIL-17F遮断の効果。Inflamm Bowel Dis. 2013;19(8):1567–1576.
この記事を経由して表示します: CrossRef PubMed Google Scholar
IL-17Aの抑制ではなくIL-17Fの抑制は、腸内細菌叢の改変を介してTreg細胞を誘導することにより、大腸炎に対する防御を提供する。Nat Immunol. 2018;19(7):755-765.
この記事を経由して見る CrossRef PubMed Google Scholar
Feng T, et al. Th17細胞は大腸炎を誘発し、自然免疫IL-12およびIL-23産生のIL-17誘導を介してTh1細胞応答を促進する。J Immunol. 2011;186(11):6313–6318.
この論文を見る CrossRef PubMed Google Scholar
RORγを発現するTh17細胞はIL-17AとIL-17Fの冗長な作用を介してマウス慢性腸炎を誘導する。Gastroenterology. 2009;136(1):257-267.
この記事を見る CrossRef PubMed Google Scholar
中等症から重症のクローン病に対するヒト抗IL-17Aモノクローナル抗体Secukinumab:無作為化二重盲検プラセボ対照試験の予期せぬ結果。Gut. 2012;61(12):1693–1700.
この記事を経由して表示します: CrossRef PubMed Google Scholar
ROR-γtの一過性の阻害は、TH17細胞を減少させ、3群自然リンパ球を温存することにより、治療的に腸の炎症を制限する。Nat Med. 2016;22(3):319-323.
この記事を見る CrossRef PubMed Google Scholar
腸上皮細胞による抗原提示。Immunol Lett. 1999;69(1):7-11.
この記事を経由して表示します: クロスリーフ PubMed Google Scholar
IFNγを介したIL-18による腸管上皮MHCIIの遺伝的および通性誘導。Mucosal Immunol. 2021;14(5):1100–1112.
この記事を見る CrossRef PubMed Google Scholar
Toll様受容体による常在細菌叢の認識は腸の恒常性維持に必要である。Cell. 2004;118:229-241.
この記事を見る クロスリーフ PubMed Google Scholar
Rhee SH, et al. 大腸炎症における細菌フラジェリンによるToll様受容体5の関与の病態生理学的役割。Proc Natl Acad Sci U S A. 2005;102(38):13610-13615.
この記事を経由して表示します: クロスリーフ PubMed Google Scholar
Rose WAI, et al. TLR9は腸管障害からの保護と腸管修復に重要である。Sci Rep.
この論文を見る PubMed Google Scholar
NF-κBを介する炎症における新たな創薬標的としてのヒストン脱アセチル化酵素3(hdac 3)。Curr Opin Chem Biol.
この記事を見る PubMed Google Scholar
Zhu H, et al. ヒストン脱アセチル化酵素3の活性化は、リポ多糖刺激時の心筋細胞における腫瘍壊死因子α(TNFα)の発現を促進する。J Biol Chem. 2010;285(13):9429–9436.
この記事を見る クロスリーフ PubMed Google Scholar
ヒストン脱アセチル化酵素は伸長機構を介して転写を正に制御する。Cell Rep. 2015;13(7):1444-1455.
この記事を見る CrossRef PubMed Google Scholar
ヒストン脱アセチル化酵素3は、トランスフォーミング成長因子β1(TGF-β1)の脱アセチル化酵素非依存的エピジェネティックサイレンシングを調整し、第二心野形成を制御する。J Biol Chem. 2015;290(45):27067–27089.
この記事を見る クロスリーフ PubMed Google Scholar
ヒストン脱アセチル化酵素3は褐色脂肪組織を急性熱発生チャレンジに備える。Nature. 2017;546(7659):544–548.
この論文を見る CrossRef PubMed Google Scholar
腸内細菌叢はNFIL3と概日時計を介して体組成を制御する。Science. 2017;916(357):912–916.
この論文を見る PubMed Google Scholar
ケラチノサイト内在性MHCII発現は微生物が誘導するTh1細胞応答を制御する。Proc Natl Acad Sci U S A. 2019;116(47):23643-23652.
経由でこの記事を見る: CrossRef PubMed Google Scholar
Kerdidani D, et al. 肺腫瘍MHCII免疫は線維芽細胞によるin situ抗原提示に依存する。J Exp Med. 2022;219(2):e20210815.
この記事を見る CrossRef PubMed Google Scholar
肺上皮細胞による抗原提示はCD4+ TRM細胞の機能を指令し、バリア免疫を制御する。Nat Commun. 2021;12(1):5834.
この論文を見る クロスリーフ PubMed Google Scholar
ヒストン脱アセチル化酵素3による概日リズムが肝脂質代謝を制御している。Science. 2011;331(6022):1315–1319.
この記事を見る クロスリーフ PubMed Google Scholar
マウスにおけるヒストン脱アセチル化酵素3による心臓エネルギー代謝の維持。J Clin Invest. 2008;118(11):3588–3597.
この記事を見る JCI CrossRef PubMed Google Scholar
固形臓器傷害の病態におけるヒストン脱アセチル化酵素3の重要な役割。Cell Death Dis. 2021;12(8):1-13.
この記事を見る PubMed Google Scholar
カンジダ・アルビカンス(Candida albicans)の腸管コロニー形成に伴うUME6発現の変動は、全身のTh17防御免疫を促進する。Cell Rep.
この記事を見る クロスリファレンス PubMed Google Scholar
Igyártó BZ, et al. 皮膚に常在するマウス樹状細胞サブセットは抗原特異的Tヘルパー細胞応答を促進する。Immunity. 2011;35(2):260-272.
この記事を見る CrossRef PubMed Google Scholar
レチノイン酸は腸内感染に対する宿主の防御を促進する。Cell Host Microbe. 2021;29(12):1744–1756.
この記事を見る クロスリーフ PubMed Google Scholar
ウッドDE、サルツバーグSL. ゲノム生物学(Genome Biol. ゲノム生物学2014;15(3):10.
この記事を見る PubMedグーグルシュラー
Vegan: コミュニティエコロジーパッケージ。Rパッケージバージョン2。Oksanen AJ, et al. 2022; https://cran.r-project.org/web/packages/vegan/index.html.
細菌フラジェリンはクローン病の支配的抗原である。J Clin Invest. 2004;113(9):1296–1306.
この記事を見る JCI CrossRef PubMed Google Scholar
Buchfink B, Reuter K, Drost HG. DIAMONDを用いたTree-of-Lifeスケールでの高感度タンパク質アラインメント。Nat Methods. 2021;18(4):366-368.
この記事を見る 論文リスト
バージョン履歴
バージョン1(2023年1月5日): インプレスプレビュー
バージョン2 (2023年2月15日): 電子出版
われわれは次のことを推奨する。
細菌フラジェリンはクローン病の支配的抗原である
マイケル・J・ロデスら、J Clin Invest誌、2004年
口腔内常在細菌叢はマウスにおいて全身性マイクロバイオームとは別に骨免疫調節作用を誘導する
Jessica D. Hathaway-Schraderら、JCI Insight、2022年
クローン病の炎症を予測する微生物叢感受性のエピジェネティックシグネチャー
ダニエル・ケリーら、JCI Insight、2018年
子牛における腸内細菌叢のコロニー形成と発達
Yufeng Duら、Journal of Animal Science and Biotechnology誌、2023年
歯周炎は唾液中の微生物叢を介して腸内細菌叢の異常を誘発する可能性がある
Jun Baoら、International Journal of Oral Science誌、2022年
ブタとブロイラーにおける熱ストレス:腸肝軸の障害における腸内細菌異常症の役割とプロバイオティクス、プレバイオティクスおよびシンバイオティクスによるこれらの効果の回復
ロバート・リングゼイスら、動物科学・バイオテクノロジージャーナル、2022年
提供
著作権 © 2023 米国臨床試験学会
ISSN: 0021-9738(印刷), 1558-8238(オンライン)
アラートメールに登録する
この記事が気に入ったらサポートをしてみませんか?