
【ATAC-seq】超絶初心者が公共データベースからATAC-seq解析にトライする

今回はATAC-seqにトライしましたので、そのメモです。
主に参考にしたサイトは下記です。
①公共データベースからデータを探す
今回は野生型のPGP1細胞のATAC-seqデータが欲しかったので、ChIP-Atlasや
NCBI GEO (Gene Expression Omnibus)からデータを探します。
NCBI-GEOでよさげなデータを見つけます。
下図のように検索欄があるので、キーワードを入れて検索します。今回は「PGP1, ATAC」と入力。

検索すると、いくつかのデータベースを見つけることができました。これをクリックします。
*「GM23338」というラベルは、今回の解析しようとしているPGP1細胞の別名です。

下の方に「Samples」と書かれたところに、この研究で使われた全サンプルが表示されます。そのうちの1つをクリック。
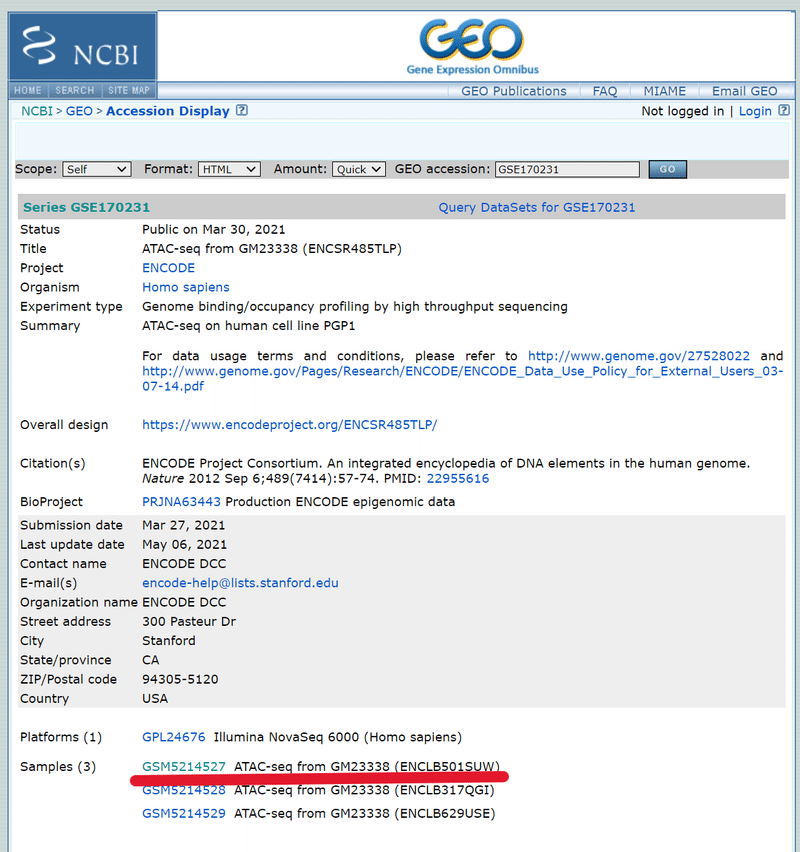
ページが切り替わり、更に下の方にスクロールすると、今度は「SRA」と書かれた部位がでてくるので、クリックする。

更にページが移動します。
このページでは、サンプルの情報が記載されています。
ちゃんと「ATAC-seq」であること、Layoutがペアエンドかどうか等を確認し、下の方にある「SRR」から始まるIDをメモしておきます。

*ChIP-seqやRNA-seqのデータと取り違える可能性があるので注意してください
②SRAファイルのダウンロード
次にSRA toolkit の「fastq-dump」コマンドを利用して FASTQ を抽出する。
fastq-dump は、NCBIのSequence Read Archive (SRA) Toolkitの一部であるコマンドラインツール。このツールは、SRAデータベースに格納されているシーケンシングデータを、一般的に使用されるFASTQ形式に変換してダウンロードするために使用される。
a) SRA toolkitのインストール
下記のコマンドを入力して、SRA toolkitのインストールし、解凍およびパスを通す。
#SRA -toolkitのインストール
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
#解凍
tar xvzf sratoolkit.current-ubuntu64.tar.gz
#解凍したファイルの移動 (verによってsratoolのファイル名が異なるため注意)
sudo mv sratoolkit.3.0.10-ubuntu64 /usr/local/src/
#パスを通す (verによってsratoolのファイル名が異なるため注意)
sudo ln -s /usr/local/src/sratoolkit.3.0.10-ubuntu64/bin/* /usr/local/bin/b) fastq-dumpを使用してgzip圧縮ファイルを取得する
下記コマンドで直接gzip形式で圧縮ファイルを作成する。
*すごく容量の大きいファイルがダウンロードされます。
#SRAファイル (ペアエンド)をgzip圧縮して直接ダウンロード
fastq-dump --gzip --split-files SRR14104174③Trim Galoreを用いたトリミング
次にトリミングを行います。
Trim Galore!のインストールに関しては、下記ページに記載しております。
a) Trim Galore!のインストール
まずはconda経由でTrim Galore!をインストールする。
#conda経由でTrimGalore!のインストール
conda install trim-galoreb) cutadaptおよびFastQCのインストール
次に、brew経由でcutadaptとFastQCをインストールする。これらのアプリがないと、Trim Galoreが作動できない。
brew install cutadapt
brew install FastQCc) Trim Galore!を用いたトリミング
上記をインストールし、Trim Galoreのパスを通したら、実際にトリミングを行う。
まずはトリミングを行いたいファイルが存在するディレクトリに移動する。そのフォルダ内で③で取得したgzip圧縮されたペアエンドfastqファイルに対して、下記のコマンドを実行する。
*なかなかに時間がかかるので注意!!
trim_galore --paired SRR14104174_1.fastq.gz SRR14104174_2.fastq.gz
④FastQCを用いてトリミングファイルのクオリティチェック
a) FastQCを用いてクオリティチェック(QC)を行う
できあがったトリミングファイルをFastQCを用いてクオリティチェックします。下記のコマンドを入力してラン。
fastqc --nogroup -o . SRR14104174_1_val_1.fq.gz
fastqc --nogroup -o . SRR14104174_2_val_2.fq.gz"--nogroup":3'末端のリードについてもちゃんと解析。*リードが長くなると、3' 末端は十数塩基がまとめて解析され、3' 末端の解析結果が曖昧となることがあるため。
SRR14104174_1_val_1.htmlというファイルができるので、ブラウザでQCの結果を確認することができる。
b) QC結果をブラウザで確認
出来上がったファイルをクリックすることで、ブラウザでQC結果を確認することができる。
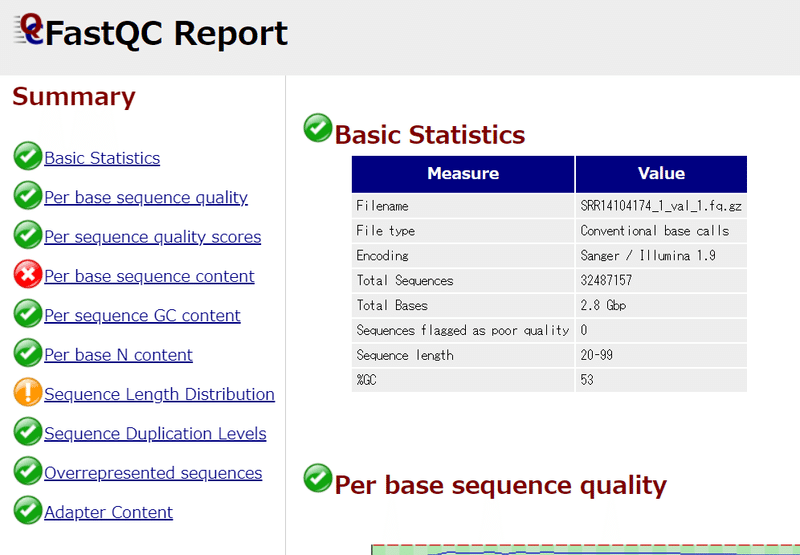
左のSummary欄がほとんど緑色の✔になればOK。
*今回は「Per base sequence content」が赤く×になっているが、サンプル処理の過程でDNaseIを使っているためと考えられる。
**FastQCに関する細かい解釈に関しては、下記サイトで確認可能。
⑤Bowtie2を用いてマッピング
a) Bowtie2のインストール
下記コマンドを入力して、brew経由でBawtie2をインストールする。
brew install bowtie2b) リファレンスゲノムのダウンロード
次に、UCSC Genome Browserからリファレンスとなるヒトゲノムデータをアダウンロードします。
*ダウンロードできない場合は、UCSCのサイトから直接ダウンロードする。
mkdir -p ~/bowtie2_index/bowtie2_human
cd bowtie2_index/bowtie2_human/
wget ftps://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.chromFa.tar.gz
tar -xzf hg38.chromFa.tar.gz次に「random」や「unknown」配列などを切り捨て、残ったものを1つのファイルに再構成し、「human_rev.fa」というファイルで保存します。
cd -
cd bowtie2_index/bowtie2_human/chroms/
rm *random.fa
rm chrUn*
rm chrM.fa
cat *.fa > human_rev.fac) インデックスの作成
いよいよインデックスを作成します。
先ほど作成した「human_rev.fa」が存在するディレクトリに移動します。
下記コマンドを実施。30分くらいかかります。
cd -
cd bowtie2_index/bowtie2_human/chroms/
mkdir human_rev_index
bowtie2-build --threads 4 -f ./chroms/human_rev.fa ./human_rev_index/human_rev_index*コマンドの説明*
"bowtie2-build" コマンドは、Bowtie2インデックスを作成します。
"--threads 4" オプションは、インデックス作成プロセスに4つのスレッド(コア)を使用することを意味し、処理を高速化します。
"-f" オプションは、入力ファイルがFASTA形式であることを指定します。
"./chroms/human_rev.fa" は、インデックスを作成するためのリファレンスゲノムのFASTAファイルのパスです。
"./human_rev_index/human_rev_index" は、作成されるインデックスファイルの出力先と名前を指定します。
実行後は、下図のよう6つのファイルが出来上がります。

d) マッピングの実施
次に、作成したインデックスを用いて、トリミングしたサンプルデータをマッピングします。
ランを実行する前に、下図の様に作成したインデックスファイルとサンプルデータが同じディレクトリに存在するように整理します。今回は「bowtie2_index/bowtie2_human」のディレクトリ内に置きました。

整理したら、下記のコマンドを実行します。
30-40分かかります。
cd -
cd bowtie2_index/bowtie2_human
bowtie2 -p 4 -x ./human_rev_index/human_rev_index -1 SRR14104174_1_val_1.fq.gz -2 SRR14104174_2_val_2.fq.gz -S SRR14104174.sam*コマンドの説明*
”bowtie2” コマンドは、リードのアライメントを実行します。
”-p 4” オプションは、アライメントプロセスに4つのスレッド(コア)を使用することを意味し、処理を高速化します。
”-x ./human_rev_index/human_rev_index” は、アライメントに使用するインデックスの場所と名前を指定します。ここでは、human_rev_index という名前のインデックスを使用しています。
”-1 SRR14104174_1_val_1.fq.gz” と ”-2 SRR14104174_2_val_2.fq.gz” は、ペアエンドリードのファイルを指定します。これらのファイルは、シーケンシング実験から得られたリードデータを含んでいます。
”-S SRR14104174.sam” は、アライメントの結果をSAM形式で出力するファイルを指定します。
できあがったファイルが下図。もの凄く大きいファイル容量…。

e) SAM → BAMファイルへの変換
出来上がったSAMファイルをsamtoolを使ってBAMファイルへ変換します。
samtools view -b -o SRR14104174.bam SRR14104174.sam*コマンドの説明*
"samtools view" は、SAM/BAM ファイル形式の変換やフィルタリングに使用される samtools のコマンドです。
"-b" オプションは、出力を BAM 形式で生成することを指示します。
5-10分程でBAMファイルへの変換が終了します。
マッピングはこれで終わりです。

⑥bigWigファイルを作成する
a) deepToolsのインストール
まず「deepTools」をconda経由でインストールします。
conda install deeptoolsb) bam.baiファイルの作成
次にsamtoolを使用して、まずはBAMファイルをソートします(特定の順番に並び変えます)。その後、BAMのインデックスファイル(bam.bai)を作成します。
*よく分かっていませんが…bigWigファイルに変換するために必要な工程らしい。
samtools sort SRR14104174.bam -o SRR14104174_sorted.bam
samtools index SRR14104174_sorted.bam
c) bigWigファイルの作成
続いて、bamCoverageを実行してbigWIgファイルを作成する。
*実行の前にsorted.bamファイルとbam.baiファイルが同じディレクトリに存在するようにする。
bamCoverage -b SRR14104174_sorted.bam -p 4 --normalizeUsing RPGC --effectiveGenomeSize 2913022398 --binSize 1 -o SRR14104174.bigwig*コマンドの解説*
"-b" オプションは入力として使用するBAMファイルを指定します。この例では、SRR14104174_sorted.bam が入力ファイルです。
"-p" オプションは使用するプロセッサ(CPUコア)の数を指定します。ここでは、4つのコアを使用します。
"--normalizeUsing" オプションは、カバレッジデータの正規化方法を指定します。RPGC はリード数をゲノムサイズで割って正規化する方法です(解析方法によって正規化の種類は変わる!)。
"--effectiveGenomeSize" オプションは、使用されるリファレンスゲノムの有効サイズ(ヌクレオチド数)を指定します。この数値は、使用するリファレンスゲノムに基づいて選ばれます(*リファレンスによる数値の違いはこのサイトを確認)。
"--binSize" オプションは、カバレッジを計算する際のビン(区間)のサイズをヌクレオチド単位で指定します。この例では、1ヌクレオチドごとのカバレッジが計算されます。
"-o" オプションは出力ファイルの名前を指定します。ここでは、出力ファイルは SRR14104174.bigwig として保存されます。
*正規化の主な種類*
RPKM/FPKM(Reads/Fragments Per Kilobase of transcript per Million mapped reads):
遺伝子の長さとシーケンスされたリードの総数に基づいて正規化します。
トランスクリプトームデータ解析や、リードの数が異なるサンプル間の比較に適しています。
TPM(Transcripts Per Million):
RPKM/FPKMに似ていますが、より高度な正規化を行います。
トランスクリプトの相対的な発現量を比較する際に有用です。
BPM(Bins Per Million):
シーケンスされたリードの総数に基づいて、各ビン(特定のゲノム領域)内のリードを正規化します。
RPGC(Reads Per Genomic Content):
シーケンスされたリードの総数とリファレンスゲノムのサイズに基づいて正規化します。
ゲノム全体のシーケンシングデータ(例:ChIP-seq)に適しています。
**正規化方法の選択**
データの種類:
・RNA-seqデータの場合、RPKMまたはTPMが一般的です。
・ChIP-seqやATAC-seqなどのゲノムワイドなデータの場合、RPGCまたはBPMが適しています。
⑦IGVで可視化する
a) IGVのインストール
下記のサイトから、自分のPCに合ったIGV (Integrative Genomics Viewer)をインストールします。IGVを使用することで、ATAC-seqやChIP-seqのシークエンス結果を可視化することができます。
b) IGVにbigWigデータを取り込む
次にデータを取り込みます。
その前に、まずIGVを立ち上げます。
立ち上げたら、下図の赤枠をクリックして、リファレンスデータとなる「Human h38」をダウンロードします。


ダウンロードが終わったら、作成したbigWigファイルをドラッグ&ドロップして取り込ませます。
すると、下図のようなデータが表示されました。

…キリが悪いですが、今回は一旦ここで終了です!
この記事が気に入ったらサポートをしてみませんか?