SARS-CoV-2ゲノムの系統発生ネットワーク分析

PNAS(米国科学アカデミー)が新型コロナウィルスを分析した結果、新型コロナウィルスが複数存在する事が判明しました。
Phylogenetic network analysis of SARS-CoV-2 genomes
SARS-CoV-2ゲノムの系統発生ネットワーク分析
Peter Forster, Lucy Forster, Colin Renfrew, and
View ORCID Profile
Michael Forster
PNAS first published April 8, 2020 https://doi.org/10.1073/pnas.2004999117
Contributed by Colin Renfrew, March 30, 2020 (sent for review March 17, 2020; reviewed by Toomas Kivisild and Carol Stocking)
ピーターフォースター、ルーシーフォースター、コリンレンフルー、そして
ORCIDプロフィールを表示
マイケル・フォースター
PNASが最初に公開されたのは2020年4月8日https://doi.org/10.1073/pnas.2004999117
2020年3月30日、Colin Renfrewによる寄稿(2020年3月17日のレビュー用に送信。ToomasKivisildとCarol Stockingによるレビュー)
Significance
This is a phylogenetic network of SARS-CoV-2 genomes sampled from across the world. These genomes are closely related and under evolutionary selection in their human hosts, sometimes with parallel evolution events, that is, the same virus mutation emerges in two different human hosts. This makes character-based phylogenetic networks the method of choice for reconstructing their evolutionary paths and their ancestral genome in the human host. The network method has been used in around 10,000 phylogenetic studies of diverse organisms, and is mostly known for reconstructing the prehistoric population movements of humans and for ecological studies, but is less commonly employed in the field of virology.
意義
これは、世界中からサンプリングされたSARS-CoV-2ゲノムの系統発生ネットワークです。 これらのゲノムは密接に関連しており、それらのヒト宿主における進化的選択の下で、時には並行進化事象を伴います。つまり、同じウイルス変異が2つの異なるヒト宿主に出現します。 これにより、キャラクターベースの系統発生ネットワークが、進化の経路と人間の宿主における祖先のゲノムを再構築するための最適な方法になります。 ネットワーク手法は、多様な生物の約10,000系統の研究で使用されており、人間の先史時代の個体群の動きの再構築や生態学の研究でよく知られていますが、ウイルス学の分野ではあまり一般的ではありません。
Abstract
In a phylogenetic network analysis of 160 complete human severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) genomes, we find three central variants distinguished by amino acid changes, which we have named A, B, and C, with A being the ancestral type according to the bat outgroup coronavirus. The A and C types are found in significant proportions outside East Asia, that is, in Europeans and Americans. In contrast, the B type is the most common type in East Asia, and its ancestral genome appears not to have spread outside East Asia without first mutating into derived B types, pointing to founder effects or immunological or environmental resistance against this type outside Asia. The network faithfully traces routes of infections for documented coronavirus disease 2019 (COVID-19) cases, indicating that phylogenetic networks can likewise be successfully used to help trace undocumented COVID-19 infection sources, which can then be quarantined to prevent recurrent spread of the disease worldwide.
概要
160の完全なヒト重症急性呼吸器症候群コロナウイルス2(SARS-Cov-2)ゲノムの系統学的ネットワーク分析では、A、B、Cという名前のアミノ酸の変化によって区別される3つの中心的なバリアントが見つかります。コウモリのアウトグループコロナウイルスによる祖先型。 A型とC型は、東アジア以外、つまりヨーロッパ人とアメリカ人にかなりの割合で見られます。対照的に、Bタイプは東アジアで最も一般的なタイプであり、その祖先のゲノムは、最初に派生Bタイプに変異することなく、東アジア外に広がっていないようであり、アジア外でのこのタイプに対する創始者効果または免疫学的または環境耐性を示しています。ネットワークは、文書化されたコロナウイルス疾患2019(COVID-19)のケースの感染経路を忠実に追跡します。系統発生ネットワークが同様に、文書化されていないCOVID-19感染源の追跡に役立ち、その後、世界的に病気の再発の拡大を防ぐために隔離できることを示しています。
The search for human origins seemed to take a step forward with the publication of the global human mitochondrial DNA tree (1). It soon turned out, however, that the tree-building method did not facilitate an unambiguous interpretation of the data. This motivated the development, in the early 1990s, of phylogenetic network methods which are capable of enabling the visualization of a multitude of optimal trees (2, 3). This network approach, based on mitochondrial and Y chromosomal data, allowed us to reconstruct the prehistoric population movements which colonized the planet (4, 5). The phylogenetic network approach from 2003 onward then found application in the reconstruction of language prehistory (6). It is now timely to apply the phylogenetic network approach to virological data to explore how this method can contribute to an understanding of coronavirus evolution.
人間の起源の探求は、世界規模の人間のミトコンドリアDNAツリーの発表と一歩前進するように見えました(1)。 しかし、すぐに、ツリー構築方法ではデータの明確な解釈が容易にならないことがわかりました。 これは、1990年代初頭に、多数の最適なツリーの視覚化を可能にすることができる系統発生ネットワーク手法の開発の動機になりました(2、3)。 このネットワークアプローチは、ミトコンドリアとY染色体のデータに基づいており、惑星に植民地化した先史時代の個体群の動きを再構築することを可能にしました(4、5)。 2003年以降の系統学的ネットワークアプローチは、言語先史時代の再構築に適用されました(6)。 この方法がコロナウイルスの進化の理解にどのように貢献できるかを調査するために、ウイルス学データに系統発生ネットワークアプローチを適用するのが今やタイムリーです。
In early March 2020, the GISAID database (https://www.gisaid.org/) contained a compilation of 253 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) complete and partial genomes contributed by clinicians and researchers from across the world since December 2019. To understand the evolution of this virus within humans, and to assist in tracing infection pathways and designing preventive strategies, we here present a phylogenetic network of 160 largely complete SARS-Cov-2 genomes (Fig. 1).
2020年3月初旬、GISAIDデータベース(https://www.gisaid.org/)には、253人の重症急性呼吸器症候群コロナウイルス2(SARS-CoV-2)の完全なゲノムと部分的なゲノムが含まれています。 このウイルスの人間内での進化を理解し、感染経路の追跡と予防戦略の設計を支援するために、ここでは160の完全なSARS-Cov-2ゲノムの系統発生ネットワークを提示します(図1)。
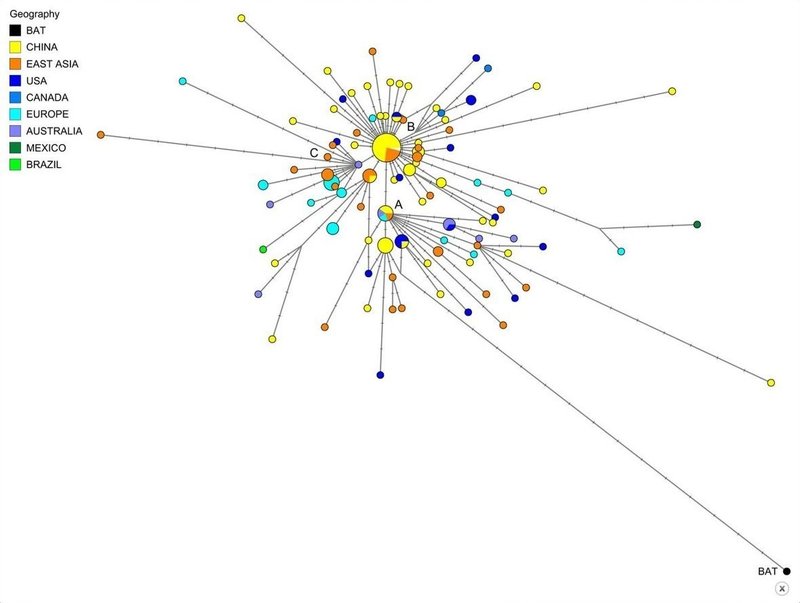
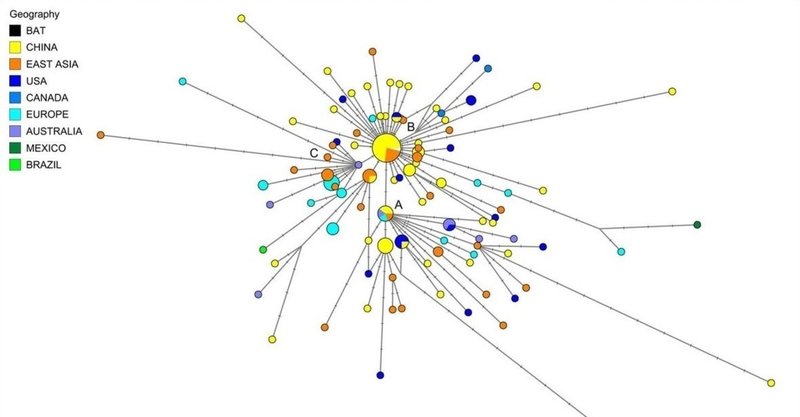
Fig. 1.
Phylogenetic network of 160 SARS-CoV-2 genomes. Node A is the root cluster obtained with the bat (R. affinis) coronavirus isolate BatCoVRaTG13 from Yunnan Province. Circle areas are proportional to the number of taxa, and each notch on the links represents a mutated nucleotide position. The sequence range under consideration is 56 to 29,797, with nucleotide position (np) numbering according to the Wuhan 1 reference sequence (8). The median-joining network algorithm (2) and the Steiner algorithm (9) were used, both implemented in the software package Network5011CS (https://www.fluxus-engineering.com/), with the parameter epsilon set to zero, generating this network containing 288 most-parsimonious trees of length 229 mutations. The reticulations are mainly caused by recurrent mutations at np11083. The 161 taxa (160 human viruses and one bat virus) yield 101 distinct genomic sequences. The phylogenetic diagram is available for detailed scrutiny in A0 poster format (SI Appendix, Fig. S5) and in the free Network download files.
図1。
160 SARS-CoV-2ゲノムの系統発生ネットワーク。ノードAは、雲南省のコウモリ(R. affinis)コロナウイルス分離株BatCoVRaTG13で取得されたルートクラスターです。円の面積は分類群の数に比例し、リンク上の各ノッチは変異したヌクレオチドの位置を表します。検討中の配列範囲は56から29,797で、ヌクレオチド位置(np)は武漢1参照配列に従って番号付けされています(8)。中央結合ネットワークアルゴリズム(2)とシュタイナーアルゴリズム(9)が使用され、どちらもソフトウェアパッケージNetwork5011CS(https://www.fluxus-engineering.com/)に実装され、パラメーターepsilonがゼロに設定されて、このネットワークには、長さが229の変異を含む最も簡潔な288本のツリーが含まれています。網状組織は主にnp11083での再発性突然変異によって引き起こされます。 161の分類群(160のヒトウイルスと1つのコウモリウイルス)は、101の異なるゲノムシーケンスを生成します。系統図は、A0ポスター形式(SI付録、図S5)および無料のネットワークダウンロードファイルで詳細に調査できます。
Zhou et al. (7) recently reported a closely related bat coronavirus, with 96.2% sequence similarity to the human virus. We use this bat virus as an outgroup, resulting in the root of the network being placed in a cluster of lineages which we have labeled “A.” Overall, the network, as expected in an ongoing outbreak, shows ancestral viral genomes existing alongside their newly mutated daughter genomes.
周他 (7)最近、ヒトウイルスと96.2%の配列類似性を持つ、密接に関連したコウモリコロナウイルスが報告されました。 このバットウイルスをアウトグループとして使用し、ネットワークのルートが、「A」というラベルが付けられた系統のクラスターに配置されます。 全体として、進行中の大発生で予想されるように、ネットワークは先祖のウイルスゲノムが新しく変異した娘ゲノムと並んで存在することを示しています。
There are two subclusters of A which are distinguished by the synonymous mutation T29095C. In the T-allele subcluster, four Chinese individuals (from the southern coastal Chinese province of Guangdong) carry the ancestral genome, while three Japanese and two American patients differ from it by a number of mutations. These American patients are reported to have had a history of residence in the presumed source of the outbreak in Wuhan. The C-allele subcluster sports relatively long mutational branches and includes five individuals from Wuhan, two of which are represented in the ancestral node, and eight other East Asians from China and adjacent countries. It is noteworthy that nearly half (15/33) of the types in this subcluster, however, are found outside East Asia, mainly in the United States and Australia.
同義変異T29095Cによって区別されるAの2つのサブクラスターがあります。 T対立遺伝子サブクラスターでは、4人の中国人(中国南部沿岸州の広東省)が祖先のゲノムを保有していますが、日本人の3人とアメリカ人の2人の患者は、多くの突然変異によって祖先ゲノムと異なります。 これらのアメリカ人患者は武漢での発生の推定原因に居住歴があったと報告されている。 C対立遺伝子サブクラスターは比較的長い突然変異の枝をスポーツし、武漢からの5人の個人が含まれ、そのうちの2つは祖先のノードで表され、他の8人の中国および隣接国からの東アジア人です。 ただし、このサブクラスターのタイプのほぼ半分(15/33)が東アジア以外の地域、主に米国とオーストラリアで見つかっていることは注目に値します。
Two derived network nodes are striking in terms of the number of individuals included in the nodal type and in mutational branches radiating from these nodes. We have labeled these phylogenetic clusters B and C.
2つの派生ネットワークノードは、ノードタイプに含まれる個体の数と、これらのノードから放射する変異ブランチで顕著です。 これらの系統発生クラスターをBおよびCとラベル付けしました。
For type B, all but 19 of the 93 type B genomes were sampled in Wuhan (n = 22), in other parts of eastern China (n = 31), and, sporadically, in adjacent Asian countries (n = 21). Outside of East Asia, 10 B-types were found in viral genomes from the United States and Canada, one in Mexico, four in France, two in Germany, and one each in Italy and Australia. Node B is derived from A by two mutations: the synonymous mutation T8782C and the nonsynonymous mutation C28144T changing a leucine to a serine. Cluster B is striking with regard to mutational branch lengths: While the ancestral B type is monopolized (26/26 genomes) by East Asians, every single (19/19) B-type genome outside of Asia has evolved mutations. This phenomenon does not appear to be due to the month-long time lag and concomitant mutation rate acting on the viral genome before it spread outside of China (Dataset S1, Supplementary Table 2). A complex founder scenario is one possibility, and a different explanation worth considering is that the ancestral Wuhan B-type virus is immunologically or environmentally adapted to a large section of the East Asian population, and may need to mutate to overcome resistance outside East Asia.
タイプBの場合、93のタイプBゲノムのうち19を除くすべてが武漢(n = 22)、中国東部の他の地域(n = 31)、および散発的に隣接するアジア諸国(n = 21)でサンプリングされました。東アジア以外では、米国とカナダのウイルスゲノムで10のBタイプが見つかりました。メキシコに1つ、フランスに4つ、ドイツに2つ、イタリアとオーストラリアにそれぞれ1つです。ノードBは、Aから2つの突然変異、すなわち同義突然変異T8782Cと非同義突然変異C28144Tによってロイシンをセリンに変更することによって派生します。クラスターBは突然変異ブランチの長さに関して印象的です:祖先のBタイプは東アジア人によって独占されていますが(26/26ゲノム)、アジア外のすべての(19/19)Bタイプゲノムは突然変異を進化させてきました。この現象は、ウイルスゲノムが中国国外に広がる前にウイルスゲノムに作用する1か月のタイムラグと付随する突然変異率によるものではないようです(データセットS1、補足表2)。複雑な創設者のシナリオは1つの可能性であり、考慮に値する別の説明は、祖先の武漢B型ウイルスが免疫学的または環境的に東アジアの人口の大部分に適応しており、東アジア外の抵抗を克服するために変異させる必要があるかもしれないということです。
Type C differs from its parent type B by the nonsynonymous mutation G26144T which changes a glycine to a valine. In the dataset, this is the major European type (n = 11), with representatives in France, Italy, Sweden, and England, and in California and Brazil. It is absent in the mainland Chinese sample, but evident in Singapore (n = 5) and also found in Hong Kong, Taiwan, and South Korea.
タイプCは、グリシンをバリンに変更する非同義変異G26144Tが親タイプBと異なります。 データセットでは、これは主要なヨーロッパのタイプ(n = 11)であり、代表はフランス、イタリア、スウェーデン、イギリス、およびカリフォルニアとブラジルにあります。 中国本土のサンプルには存在しませんが、シンガポール(n = 5)で明らかで、香港、台湾、韓国でも見られます。
One practical application of the phylogenetic network is to reconstruct infection paths where they are unknown and pose a public health risk. The following cases where the infection history is well documented may serve as illustrations (SI Appendix). On 25 February 2020, the first Brazilian was reported to have been infected following a visit to Italy, and the network algorithm reflects this with a mutational link between an Italian and his Brazilian viral genome in cluster C (SI Appendix, Fig. S1). In another case, a man from Ontario had traveled from Wuhan in central China to Guangdong in southern China and then returned to Canada, where he fell ill and was conclusively diagnosed with coronavirus disease 2019 (COVID-19) on 27 January 2020. In the phylogenetic network (SI Appendix, Fig. S2), his virus genome branches from a reconstructed ancestral node, with derived virus variants in Foshan and Shenzhen (both in Guangdong province), in agreement with his travel history. His virus genome now coexists with those of other infected North Americans (one Canadian and two Californians) who evidently share a common viral genealogy. The case of the single Mexican viral genome in the network is a documented infection diagnosed on 28 February 2020 in a Mexican traveler to Italy. Not only does the network confirm the Italian origin of the Mexican virus (SI Appendix, Fig. S3), but it also implies that this Italian virus derives from the first documented German infection on 27 January 2020 in an employee working for the Webasto company in Munich, who, in turn, had contracted the infection from a Chinese colleague in Shanghai who had received a visit by her parents from Wuhan. This viral journey from Wuhan to Mexico, lasting a month, is documented by 10 mutations in the phylogenetic network.
系統発生ネットワークの実用的なアプリケーションの1つは、感染経路が未知であり、公衆衛生上のリスクがある場所での感染経路の再構築です。感染履歴が十分に文書化されている次のケースは、例として役立つ場合があります(SI付録)。 2020年2月25日、最初のブラジル人はイタリア訪問後に感染したと報告されており、ネットワークアルゴリズムはこれを反映して、クラスターCのイタリア人と彼のブラジルのウイルスゲノム間の変異リンクを反映しています(SI付録、図S1)。別のケースでは、オンタリオ州の男性が中国中部の武漢から中国南部の広東省に旅行し、その後カナダに戻り、そこで病気になり、2020年1月27日にコロナウイルス病2019(COVID-19)と確定診断されました。系統発生ネットワーク(SI付録、図S2)、彼のウイルスゲノムは、再構成された祖先ノードから分岐し、旅行履歴と一致して、佛山と深セン(両方とも広東省)で派生したウイルスの亜種を持っています。彼のウイルスゲノムは現在、明らかに共通のウイルス系統を共有している他の感染した北米人(カナダ人1人とカリフォルニア人2人)のウイルスゲノムと共存しています。ネットワーク内の単一のメキシコのウイルスゲノムのケースは、2020年2月28日にイタリアへのメキシコ人旅行者で診断された文書化された感染症です。ネットワークはメキシコウイルスのイタリアの起源を確認するだけでなく(SI付録、図S3)、このイタリアのウイルスは2020年1月27日にドイツで最初に文書化されたドイツの感染に由来することを意味します。ミュンヘンは、武漢から両親の訪問を受けた上海の中国人の同僚から感染に感染した。 1か月続く武漢からメキシコへのこのバイラルジャーニーは、系統発生ネットワークの10の突然変異によって記録されています。
This viral network is a snapshot of the early stages of an epidemic before the phylogeny becomes obscured by subsequent migration and mutation. The question may be asked whether the rooting of the viral evolution can be achieved at this early stage by using the oldest available sampled genome as a root. As SI Appendix, Fig. S4 shows, however, the first virus genome that was sampled on 24 December 2019 already is distant from the root type according to the bat coronavirus outgroup rooting.
このウイルスのネットワークは、その後の移動と突然変異によって系統が不明瞭になる前の、伝染病の初期段階のスナップショットです。 ウイルス進化の発根は、入手可能な最も古いサンプリングされたゲノムを根として使用することによって、この初期段階で達成できるかどうかという質問がされるかもしれません。 しかし、SI付録として、図S4が示すように、2019年12月24日にサンプリングされた最初のウイルスゲノムは、コウモリコロナウイルスのアウトグループ発根によると、すでにルートタイプから離れています。
The described core mutations have been confirmed by a variety of contributing laboratories and sequencing platforms and can be considered reliable. The phylogeographic patterns in the network are potentially affected by distinctive migratory histories, founder events, and sample size. Nevertheless, it would be prudent to consider the possibility that mutational variants might modulate the clinical presentation and spread of the disease. The phylogenetic classification provided here may be used to rule out or confirm such effects when evaluating clinical and epidemiological outcomes of SARS-CoV-2 infection, and when designing treatment and, eventually, vaccines.
説明されているコア変異は、さまざまな研究室やシーケンスプラットフォームによって確認されており、信頼できると見なすことができます。 ネットワーク内の系統地理パターンは、独特の移動履歴、創始者イベント、およびサンプルサイズによって影響を受ける可能性があります。 それにもかかわらず、変異変異体が臨床症状と疾患の拡大を調節する可能性を検討することは賢明でしょう。 ここで提供される系統分類は、SARS-CoV-2感染の臨床的および疫学的結果を評価するとき、および治療、そして最終的にはワクチンを設計するときに、そのような影響を除外または確認するために使用できます。
Materials and Methods
The Global Initiative on Sharing Avian Influenza Data (GISAID) was founded in 2006, and, since 2010, has been hosted by the German Federal Ministry of Food, Agriculture and Consumer Protection. GISAID has also become a coronavirus repository since December 2019. As of 4 March 2020, the cutoff point for our phylogenetic analysis, the GISAID database (https://www.gisaid.org/) had compiled 254 coronavirus genomes, isolated from 244 humans, nine Chinese pangolins, and one bat Rhinolophus affinis (BatCoVRaTG13 from Yunnan Province, China). The sequences have been deposited by 82 laboratories listed in Dataset S1, Supplementary Table 1. Although SARS-CoV-2 is an RNA virus, the deposited sequences, by convention, are in DNA format. Our initial alignment confirmed an earlier report by Zhou et al. (7) that the pangolin coronavirus sequences are poorly conserved with respect to the human SARS-CoV-2 virus, while the bat coronavirus yielded a sequence similarity of 96.2% in our analysis, in agreement with the 96.2% published by Zhou et al. We discarded partial sequences, and used only the most complete genomes that we aligned to the full reference genome by Wu et al. (8) comprising 29,903 nucleotides. Finally, to ensure comparability, we truncated the flanks of all sequences to the consensus range 56 to 29,797, with nucleotide position numbering according to the Wuhan 1 reference sequence (8). The laboratory codes of the resulting 160 sequences and the bat coronavirus sequences are listed in Dataset S1, Supplementary Table 2 (Coronavirus Isolate Labels).
材料および方法
鳥インフルエンザデータの共有に関するグローバルイニシアチブ(GISAID)は2006年に設立され、2010年以降、ドイツ連邦政府の食品、農業、消費者保護省によって主催されています。 GISAIDは、2019年12月からコロナウイルスリポジトリにもなりました。2020年3月4日の時点で、系統解析のカットオフポイントであるGISAIDデータベース(https://www.gisaid.org/)は、244人の人間から分離された254個のコロナウイルスゲノムをコンパイルしました、9つの中国のセンザンコウ、1つのコウモリRhinolophus affinis(中国、雲南省のBatCoVRaTG13)。配列はデータセットS1、補足表1にリストされている82の研究所によって寄託されています。SARS-CoV-2はRNAウイルスですが、寄託された配列は慣例によりDNA形式です。私たちの最初の調整は、Zhouらによる以前の報告を裏付けました。 (7)センザンコウコロナウイルスの配列は、ヒトSARS-CoV-2ウイルスに関して保存性が低いが、コウモリコロナウイルスは、我々の分析で96.2%の配列類似性を示しました。私たちは部分的な配列を破棄し、Wuらによって完全な参照ゲノムにアラインした最も完全なゲノムのみを使用しました。 (8)29,903ヌクレオチドを含む。最後に、比較可能性を確保するために、武漢1参照配列(8)に従ってヌクレオチド位置番号を付けて、すべての配列の側面をコンセンサス範囲56〜29,797に切り詰めました。得られた160のシーケンスとコウモリコロナウイルスシーケンスの実験室コードは、データセットS1、補足表2(コロナウイルス分離ラベル)にリストされています。
The 160 human coronavirus sequences comprised exactly 100 different types. We added to the data the bat coronavirus as an outgroup to determine the root within the phylogeny. Phylogenetic network analyses were performed with the Network 5011CS package, which includes, among other algorithms, the median joining network algorithm (3) and a Steiner tree algorithm to identify most-parsimonious trees within complex networks (9). We coded gaps of adjacent nucleotides as single deletion events (these deletions being rare, up to 24 nucleotides long, and mostly in the amino acid reading frame) and ran the data with the epsilon parameter set to zero, and performed an exploratory run by setting the epsilon parameter to 10. Both settings yielded a low-complexity network. The Steiner tree algorithm was then run on both networks and provided the identical result that the most-parsimonious trees within the network were of length 229 mutations. The structures of both networks were very similar, with the epsilon 10 setting providing an additional rectangle between the A and B clusters. The network output was annotated using the Network Publisher option to indicate geographic regions, sample collection times, and cluster nomenclature.
160のヒトコロナウイルスシーケンスは、100の異なるタイプで構成されていました。系統内の根を決定するために、コウモリコロナウイルスをアウトグループとしてデータに追加しました。系統発生ネットワーク分析は、ネットワーク5011CSパッケージで実行されました。これには、他のアルゴリズムの中でも、中央結合ネットワークアルゴリズム(3)とシュタイナーツリーアルゴリズムが含まれており、複雑なネットワーク(9)内の最も節約的なツリーを識別します。隣接するヌクレオチドのギャップを単一の削除イベントとしてコード化し(これらの削除はまれであり、長さが最大24ヌクレオチドで、主にアミノ酸のリーディングフレームにあります)、イプシロンパラメーターをゼロに設定してデータを実行し、設定によって探索的実行を実行しましたepsilonパラメータを10に設定します。どちらの設定でも複雑度の低いネットワークが生成されました。次に、シュタイナーツリーアルゴリズムが両方のネットワークで実行され、ネットワーク内の最も節約的なツリーの長さが229変異であるという同じ結果が得られました。両方のネットワークの構造は非常に似ており、イプシロン10の設定により、AクラスターとBクラスターの間に追加の長方形が提供されました。ネットワーク出力には、ネットワークパブリッシャーオプションを使用して注釈が付けられ、地理的な地域、サンプル収集時間、クラスターの命名法が示されています。
Data Availability.
The nucleotide sequences of the SARS-CoV-2 genomes used in this analysis are available, upon free registration, from the GISAID database (https://www.gisaid.org/). The Network5011 software package and coronavirus network files are available as shareware on the Fluxus Technology website (https://www.fluxus-engineering.com/).
データの可用性。
この分析で使用されるSARS-CoV-2ゲノムのヌクレオチド配列は、無料の登録時にGISAIDデータベース(https://www.gisaid.org/)から入手できます。 Network5011ソフトウェアパッケージとコロナウイルスネットワークファイルは、フルクサステクノロジーのWebサイト(https://www.fluxus-engineering.com/)でシェアウェアとして入手できます。
Acknowledgments
We gratefully acknowledge the authors and originating and submitting laboratories of the sequences from GISAID’s EpiFlu(TM) Database on which this research is based. We are grateful to Trevor Bedford (GISAID) for providing instructions and advice on the database. A table of the contributors is available in Dataset S1, Supplementary Table 1. We thank Arne Röhl for assessing the network.
謝辞
著者らは、この研究の基礎となっているGISAIDのEpiFlu(TM)データベースからのシーケンスの作成者と作成および提出ラボに感謝します。 データベースに関する指示とアドバイスを提供してくれたTrevor Bedford(GISAID)に感謝します。 貢献者の表は、データセットS1の補足表1にあります。ネットワークの評価を行ってくれたArneRöhlに感謝します。
引用終わり
要するに今のところコロナウィルスは3種類あるっていうのは理解できた。
この記事が気に入ったらサポートをしてみませんか?