
Nishikaコンペ:材料の物性予測 ~機械学習で材料の研究開発を推進しよう~その1(チュートリアルの理解)

1.概要
Nishikaで2023/03/10まで開催中のコンペ「材料の物性予測 ~機械学習で材料の研究開発を推進しよう~」に参加してみました。
専門ではないのですが将来的に使う可能性があるスキルのため、試行錯誤しながら少しだけ頑張ってみます。
本記事では「Nishikaが公開しているチュートリアルを見ながらコンペを理解する」ことを紹介します。オリジナルの内容は結果がでたら記事にします。
2.データの理解
2-1.データ概要の確認
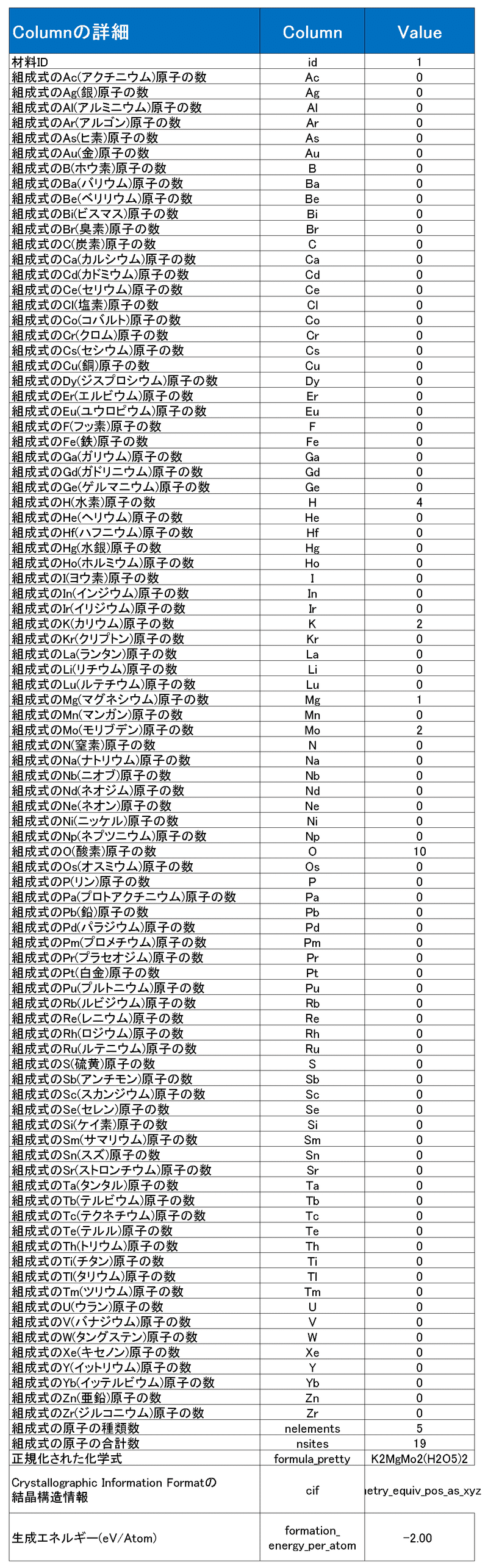
データは化合物の物性・組成情報から「生成エネルギー(eV/atom):標準状態の元素に対する化合物のエネルギーを原子単位に正規化した値」を求める回帰分析の問題です。参考に、1つのデータを下記に示します。
本コンペのポイントは下記の通りです。
基本的にはテーブルデータのため、数値データをGBDT(XGBoost)のような強いモデルに与えればそれなりの結果は出そう
原子数のデータはスパース(下表の通りほとんどが0の値)のため、そのまま使用するのは効率が悪そう
数値データとは別にカテゴリカルデータとして「formula_pretty」と「cif」がある
CIF(Crystallographic Information Format:結晶構造データ用のフォーマット)には与えられたテーブルデータ以上の重要な情報(格子定数・構造式など)が与えられているため、このデータをうまく使用した方がよさそう
本コンペでは「外部データは第三者の権利を侵害しない、無償で誰でも手に入るオープンなデータに限り外部データの利用が許可」のため、必要なら使っていきたい
(大学の授業をもう忘れたためうろ覚えですが、特徴エンジニアリングとして)生成エネルギー≒化合物の安定性のため、原子が持つ情報(電子配置、イオン化エネルギーなど)と化合物が持つ情報(原子間の距離、結晶構造など)を使用した方がよさそう。
公式ではモデルのCGCNN[Crystal Graph Convolutional Neural Networks]を公開しております。公式のモデルをそのまま使用してもLightGBMの方が結果は圧倒的に良かったため、モデルの適用にCGCNNを追い求めるかは検証しながら確認していく(※Torchの環境構築のためRAdam->Adamに変更したため精度がでなかった可能性もある)。
$$
評価指標:RSME=\sqrt{\frac{1}{N}\sum_{i=1}^{N}(y_i-\hat{y_i})^2}
$$
N:データ数、$${y_i}$$:正解値(ラベル)、$${\hat{y_i}}$$:推論値
外部データの入手も可能であり、参考までに公式に共有されていたURLの情報を下記にも記載しておきます。
[IN]
#参考情報
url='https://kunisan.jp/gomi/element_property_chart.html' #元素特性一覧表 - 原子量・密度・融点・沸点他
df_elemprop = pd.read_html(url)[0]
url = 'https://www.hulinks.co.jp/support/c-maker/qa_005.html' #元素、原子半径と周期表
df_radius = pd.read_html(url)[1]
display(HorizontalDisplay(df_elemprop, df_radius))
[OUT]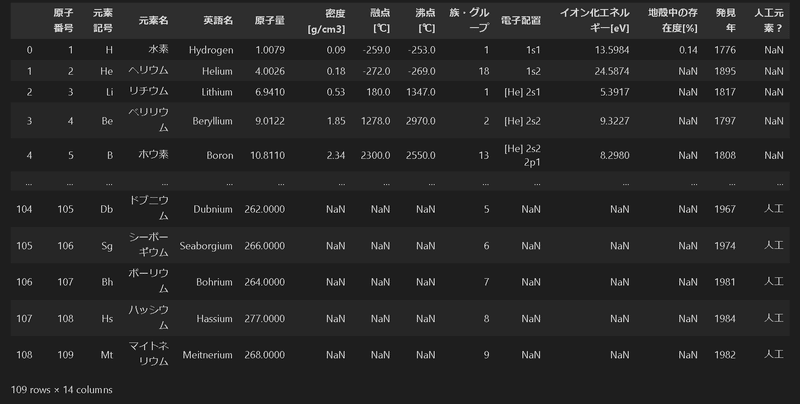

2-2.公式チュートリアルのダウンロード
Nishikaより解法のサンプルが2種類公開されております。
一つはテーブルデータ最強と呼び声が高いGBDTです。もう一つは比較的最近のモデルであるCGCNNとなります。両方ともDLして中身を確認していきます。

3.データの前処理・可視化(全般)
まずはデータの中身を確認していきます。モデルに合わせた前処理が必要となりますが、必ず実施する項目だけ本章で紹介します。
必要なライブラリのインポート、よく使うクラス・関数の作成
訓練データ・学習データ読み込み・中身の確認(PandasでDf化)
欠損値の確認
データ情報の確認(解析・可視化)
(状況に応じて)学習・訓練用のデータに分割(or 交差検証用のデータ作成)
なおNishikaのチュートリアルに合わせて「"data", "feats", "model", "output"」のフォルダを作成しました(自分用で他のフォルダも作成してます)。
3-1.ライブラリのインポート、クラス・関数作成
各種ライブラリをインポート、クラス・関数の作成をします。
[IN]
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import japanize_matplotlib
import seaborn as sns
import os
import glob
import shutil
from requests import get
import re
import itertools
from tqdm import tqdm
import missingno as msno
import random
import joblib
import pickle
import lightgbm as lgbm
from sklearn.metrics import mean_squared_error
from sklearn.model_selection import KFold
import torch
import torch.nn as nn
import torch.nn.functional as F
from torch.utils.data import Dataset, DataLoader
from torch.optim.lr_scheduler import StepLR
from pymatgen.core import Structure
class HorizontalDisplay:
def __init__(self, *args):
self.args = args
def _repr_html_(self):
template = '<div style="float: left; padding: 10px;">{}</div>'
return ''.join(template.format(a._repr_html_()) for a in self.args)
[OUT]参考までに一部のライブラリは記事を作成しているためリンクをつけました。LightGBMはXGBoostとほぼ同じためこちらで代用しています。
なお本コンペで初めて知ったライブラリのリンクを貼っておきます。
missingno:欠損値を可視化・分析する
pymatgen(Python Materials Genomics):材料解析のためのオープンソースライブラリであり、結晶構造やバンド構造などの各種物性データを簡単に取り扱うことができます。データは「Materials Project」で管理されております。
3-2.データ読み込み/確認
下記手順でデータを読み込み、中身を確認しました。
データはCSVファイルのテーブルデータのため、globでファイルパスを取得してPandasでDataFrame作成
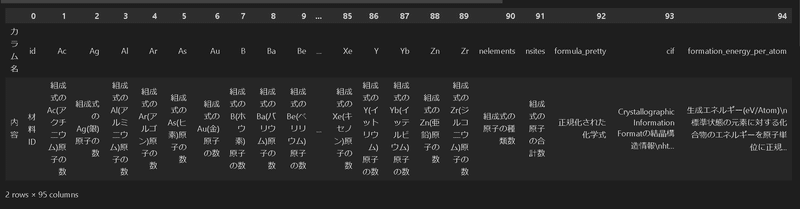
"data_explanation.xlsx"ファイルにカラム情報があるため、別途Pandasでデータ抽出して中身を確認(ファイルサイズは小さいため手動で開いてもOK)
公式にも記載されておりますが、データ情報は下記の通りです。データの読み込み時点で「値の最大値、最小値、外れ値」などの統計情報を取得しておきます。
id: 材料のid
Ac, Ag, ..., Zn, Zr: などの89種類の原子が構造式に含まれる数量
nsites: 組成式に含まれる原子の合計数
nelements: 組成式に含まれる原子の種類数
formula_pretty: 正規化された化学式
cif: Crystallographic Information File形式の結晶構造情報
CIFの詳細については参考文献[1][3]をご参照下さい。formation_energy_per_atom: 生成エネルギー(目的変数)
[IN]
#データの中身確認
datafiles = glob.glob('data/*')
file_explain, file_submit, file_test, file_train = datafiles
df_train = pd.read_csv(file_train) #訓練用データ(train.csv)
df_test = pd.read_csv(file_test) #評価用データ(test.csv)
df_submit = pd.read_csv(file_submit) #投稿データフォーマット(sample_submission.csv)
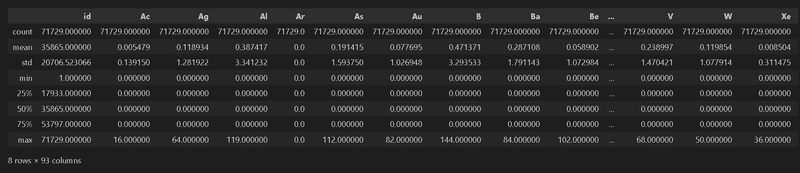
print(f'データ形状(train):{df_train.shape}、データ形状(test):{df_test.shape}、データ形状(submit):{df_submit.shape}')
display(df_train.head(3))
display(df_train.describe())
excel_pd = pd.ExcelFile(file_explain) #データ説明(data_explanation.xlsx)
sheetnames = excel_pd.sheet_names
df_explain = pd.read_excel(file_explain, sheet_name=sheetnames) #データ説明(data_explanation.xlsx)
display(df_explain['カラム説明'].T)
[OUT]
データ形状(train):(71729, 95)、データ形状(test):(28691, 94)、データ形状(submit):(28691, 2)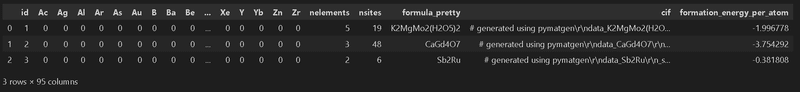
3-3.欠損値の確認・処理
テーブルデータにおける欠損値確認はPandasの下記メソッドを使用します。
df.isnull().sum():各カラムにおける欠損値の合計を集計(出力:Series型)
Series.unique():Series内のユニークな(重複が無い)データの種類を出力する(出力:List型)。
”Series.value_counts():Series内のユニークなデータの数を集計する。”df.isnull().sum()”と合わせると各欠損値数が何個あるか確認できる
結果として"formula_pretty(正規化された化学式)"カラムに1個だけエラーがあることが確認できました。本記事では"formula_pretty"カラムは入力値として使用しないため欠損値の処理はせずに進行します。
[IN]
#欠損値確認
#訓練用データ
train_Null = df_train.isnull().sum() #各項目(カラム)の欠損値の数を集計
print(f'欠損値数の種類:{train_Null.unique()}') #欠損値の個数の種類を表示
print(train_Null.value_counts()) #欠損値の個数ごとのカラムを修正
print(train_Null[train_Null!=0]) #欠損値のあるカラムを表示
#評価用データ
test_Null = df_test.isnull().sum() #テストデータには欠損値はない
print(f'欠損値の個数の種類:{test_Null.unique()}')
[OUT]
欠損値数の種類:[0 1]
0 94
1 1
dtype: int64
formula_pretty 1
dtype: int64
欠損値の個数の種類:[0]【参考:missingnoライブラリを使用】
一応使ってみましたが良くわからなかったので参考程度に乗せておきます。
[IN]
msno.matrix(df_train, figsize=(10, 5))
# msno.bar(df_train, figsize=(10, 5))
# msno.heatmap(df_train)
# msno.dendrogram(df_train, figsize=(10, 5))
[OUT]
3-4.カテゴリカルデータの数値化
数値として扱えないようなカテゴリカルデータがある場合は「One-Hotエンコーディング」や「ラベルエンコーダー」などでモデルが学習できるように数値化します。
今回のデータではカテゴリカルデータとして「formula_pretty」と「cif」のみであり、CIFは別途処理するため追加処理は不要とします。
3-5.データの解析・可視化
機械学習を実施するためにドメイン知識(所謂その分野の専門知識)を理解することは重要ですが一朝一夕で身につくものではありません。ただし、ある程度下記内容は確認しておく方がモデル作成時には便利となります。
【単変量解析(univariate analysis):要因が 1 つの場合の解析方法】
●データの分布:ヒストグラム・箱ひげ図など
●外れ値
【多変量解析(multivariate analysis):要因が 2 つ以上の場合の解析方法】
●散布図
●相関関係(相関係数)
3-5-1.データの分布1:sns.pairplot()
データ分布確認でお勧めは「Serbornのpairplot」となります。本メソッドで各説明変数$${x}$$の分布(密度分布やヒストグラム)と相関関係をまとめて出力できます。
下記に①(原子の合計数, 原子の種類数, 生成エネルギー)のプロット、②原子数の合計をラベルにして同様なプロット の2種を作成しました(最初のsns.setは記事用のための体裁調整のため通常はは無しで問題ございません)。
[IN]
sns.set(rc={'figure.figsize':(10, 10), 'figure.facecolor':'w'}) #SeabornのSetting
sns.pairplot(df_train[['nelements','nsites','formation_energy_per_atom']],
diag_kind='kde', height=3.0)
markers = ['o', 's', 'D', 'P', '*', 'X', 'v', '^']
print(df_train['nelements'].nunique())
sns.pairplot(df_train[['nelements','nsites','formation_energy_per_atom']],
hue='nelements', palette='Set2', diag_kind='kde', height=3.0,
markers=markers)
[OUT]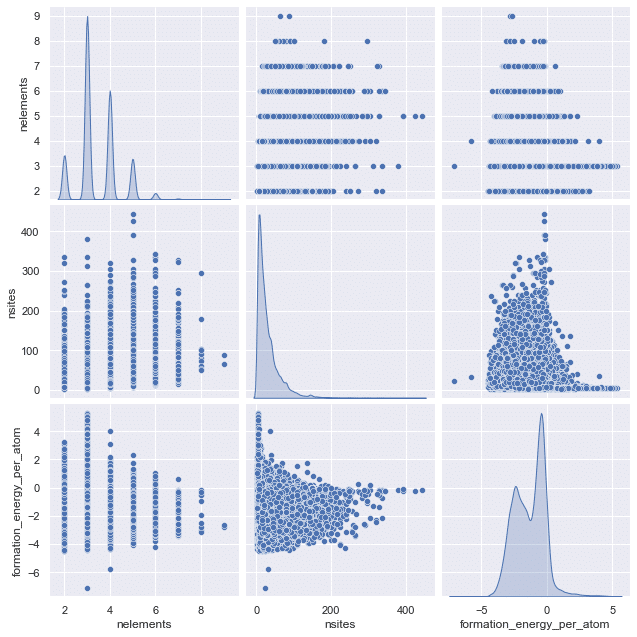
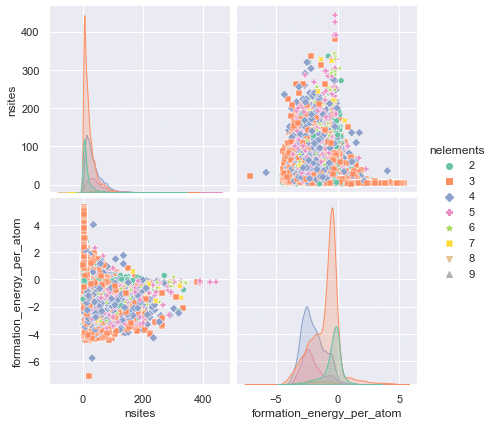
結果として下記が確認できます。
生成エネルギーに対して明確な相関がある説明変数は図では理解できない。
nelements:(組成式に含まれる原子の種類数)は離散値のためとびとびであり、ほとんどが2~6に固まっている(ほとんどの化合物は2~6種の原子で形成されている)。また最大値が9であり外れ値もない
nsites( 組成式に含まれる原子の合計数)はほとんどが100以下である。少量ではあるが100個以上の原子で形成されている化合物も存在する。なお最大値は400あたりであり一部外れ値を含む。
生成エネルギーはほとんどが0以下であり、極端な偏りない状態で分布している。
【Nishika公式の単変量解析】
Nishikaチュートリアルでの手法は下記の通りです。
[IN]
#Config:設定用データまとめのクラス
class CFG:
base_dir = "."
data_dir = os.path.join(base_dir, "data")
model_dir = os.path.join(base_dir, "model")
output_dir = os.path.join(base_dir, "output")
random_seed = 42
target_col = "formation_energy_per_atom"
n_fold = 5
cols = ['nelements', 'nsites', CFG.target_col]
colors = ['red', 'green', 'blue']
fig = plt.figure(figsize = (18, 6)) # Figureオブジェクトを作成
axs = fig.subplots(nrows = 1, ncols = 3) # Axesオブジェクトを作成
for idx, (col, color) in enumerate(zip(cols, colors)):
if col == 'nelements':
nbins = train_df[col].nunique() # カテゴリ数
else:
nbins = int(np.log2(len(train_df))) + 1 # スタージェスの公式によるビン数
axs[idx].hist(train_df[col], bins = nbins, color = color) #ヒストグラムを作成
axs[idx].set_title(f'{col}\'s histgram', y=-0.11) #タイトルを設定
plt.show()
[OUT]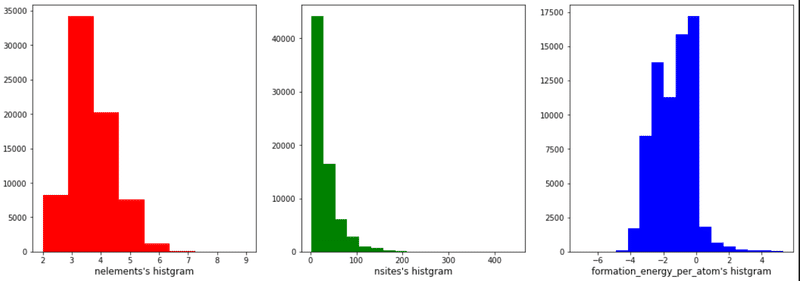
3-5-2.データの分布2:スパース性の確認
最初のモデルでは”構造式に含まれる原子の数量”を入力値として使用していく予定です(本記事のLightGBMも同様の変数を使用します。)。
今回のデータはかなり疎(スパース)なデータであり、そのまま入力値として使用するのは効率が悪そうです。とりあえず、そのくらい疎なのかを可視化しました。
可視化として①"Ac, Ag, ..., Zn, Zr"をそのまま可視化、②(最大値が大きいため)最大値1になるように正規化 したものをプロットしました。結果として下記が確認できました。
ほとんどの値が0(青色)になっています。
酸素Oは比較的多いことが確認できます(酸化物が多い)
[IN]
fig, axs = plt.subplots(1, 2, figsize=(16, 8), facecolor='white')
df_atoms = df_train.drop(columns=['nelements', 'nsites', 'formula_pretty', 'cif', 'formation_energy_per_atom']).set_index('id')
#ヒートマップで可視化
sns.heatmap(df_atoms, cmap='coolwarm', ax=axs[0])
#最大値を1に正規化
df_atoms_std = df_atoms / df_atoms.max()
df_atoms_std.fillna(0, inplace=True) #Arなど0の値のものはNullになっているので0で埋める
sns.heatmap(df_atoms_std, cmap='coolwarm', ax=axs[1])
[OUT]
3-5-3.相関係数の確認
各変数の相関が高い場合は「多重共線性による性能低下」が生じます。そのため相関を確認したうえで必要であれば次元削減や不要な変数の削除をします。今回は①原子数関係のみ、②全数値データ の相関係数を可視化します。
結果として下記の通りです。
(原子の合計数, 原子の種類数, 生成エネルギー)には強い相関が無いため、そのまま学習に使用しても問題なさそうです。(元素の種類数と生成エネルギーにやや相関がありますが、複雑(元素の種類数)との関係があるかもしれません)
基本的には各組成で相関はないが、酸素Oと水素Hで少し強めの相関が確認された。推定として、Oとnsitesの関係は原子数が多いものは酸化物で存在しているためと思います。また生成エネルギーに負の相関があるのは、もしかすると
[IN]
rename = {'nsites':'原子の合計数',
'nelements':'元素の種類数',
'formation_energy_per_atom':'原子単位の形成エネルギー'}
japanize_matplotlib.japanize() #日本語化
df_corr = df_train[["nelements", "nsites", "formation_energy_per_atom"]].corr() #相関係数の算出
display(df_corr)
#ヒートマップで可視化
plt.figure(figsize=(10, 10))
sns.heatmap(df_corr.rename(columns=rename, index=rename),
annot=True, cmap='coolwarm')
[OUT]
[IN]
sns.set(rc={'figure.figsize':(19, 19)})
sns.heatmap(df_train.drop(columns=['id', 'formula_pretty', 'cif']).corr(),
cmap='coolwarm', annot=False, cbar=True)
[OUT]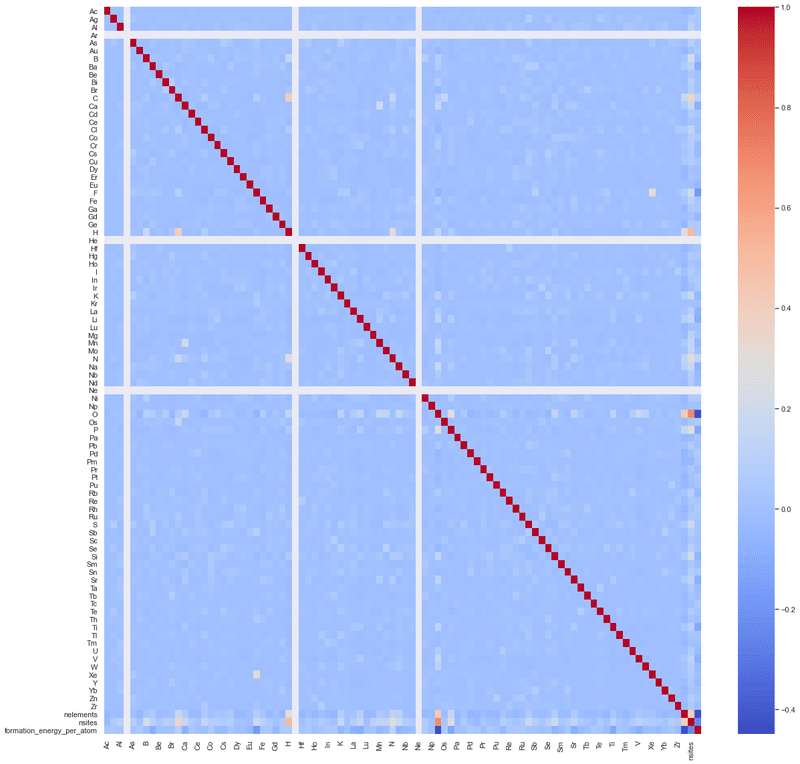
3-6.データの分割
こちらは次章以降で交差検証を実施予定のため本節では実施しません。
[IN]
[OUT]4.モデル実装1:LightGBM(GBDT)
Nishikaより「データとして与えられている組成式を構成する原子の数と原子の集約特徴量(組成式の記述子※)を使用してLightGBMで学習・推論」を行います。(出典:無機材料の組成式を元にした物性予測のための記述子開発)
つまり説明変数$${x}$$は下記2パターンでモデルを作成します(推論はCase2のみ)。
Case1:元素数情報のみを使用
Case2:元素数情報、原子の合計数(nsites)、原子の種類数(nelements)を使用
4-1.K-分割交差検証:KFold()
単純なデータ分割だけ(ホールドアウト法)だと”分割したデータに機械学習が上手くはまってたまたま性能が出た(過学習)”という可能性があるため、n等分したうえで訓練:$${\frac{n-1}{n}}$$、検証:$${\frac{1}{n}}$$で学習を平均化させます。この手法を交差検証法と呼びます。

交差検証はKFold()で実装できます。まずは簡単な動作確認を実施します。下記の通り検証データは各foldで重複せず出力はIndexとなります。
[IN]
sampledata = np.arange(15)
kf = KFold(n_splits=5, random_state=42, shuffle=True) #KFoldの設定
for train_idx, val_idx in kf.split(sampledata):
print(f'訓練データ: {train_idx}', f'検証データ: {val_idx}')
print(train_idx.shape, val_idx.shape)
[OUT]
訓練データ: [ 1 2 3 4 5 6 7 8 10 12 13 14] 検証データ: [ 0 9 11]
(12,) (3,)
訓練データ: [ 0 1 2 3 4 6 7 9 10 11 12 14] 検証データ: [ 5 8 13]
(12,) (3,)
訓練データ: [ 0 3 4 5 6 7 8 9 10 11 12 13] 検証データ: [ 1 2 14]
(12,) (3,)
訓練データ: [ 0 1 2 3 5 6 8 9 11 12 13 14] 検証データ: [ 4 7 10]
(12,) (3,)
訓練データ: [ 0 1 2 4 5 7 8 9 10 11 13 14] 検証データ: [ 3 6 12]
(12,) (3,)今回は5分割にする予定です。各foldにおける検証データの番号振りを下記コードで実行しました。下記の通りラベリング0~4でデータが等分割に分けられました。
[IN]
kf = KFold(n_splits=5, random_state=42, shuffle=True) #KFoldの設定
targets = df_train['formation_energy_per_atom'].values #目的変数(Numpy配列に変換)
#DataFrameに交差検証時のfold番号(何番目か)を追加
for i, (train_idx, val_idx) in enumerate(kf.split(targets)):
df_train.loc[val_idx, 'val_fold'] = i #foldの列にfold番号を格納
df_train['val_fold'].value_counts() #foldの分布を確認
[OUT]
1.0 14346
2.0 14346
0.0 14346
3.0 14346
4.0 14345
Name: val_fold, dtype: int64公式ではさらに検証後のデータ分布に偏りが発生していないかも確認しています(無作為抽出により偏りが発生していないことを確認)。
[IN]
#各foldの目的変数の分布を確認
fig = plt.figure(figsize = (25, 6))
axs = fig.subplots(nrows = 1, ncols = 5) # 5cols
nbins = int(np.log2(len(df_train)) + 1) # Sturges' formula
colors = ['red', 'green', 'blue', 'yellow', 'black']
#各fold(0~4)におけるデータをヒストグラムで可視化
for idx, color in enumerate(colors):
values = df_train.loc[df_train['val_fold'] == idx, 'formation_energy_per_atom'].to_numpy() #fold番号がidxのデータのみ抽出
axs[idx].hist(values, bins = nbins, color = color) #ヒストグラムの作成
axs[idx].set_title(f'val_fold:{idx}, formation_energy_per_atom\'s histgram') #タイトルの設定
plt.show()
[OUT]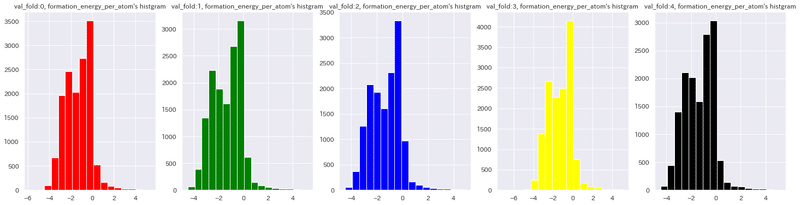
4-2.特徴量(記述子)の作成
前述の通り下記2種の特徴量でモデルを学習させます。なお公式では”記述子(descriptor)※”という表現が使用されておりますが、おそらく説明変数と同様の意味だと思います。
(※明確な記載がないため分かりにくいですが、おそらく物理定数や原子定数のような因果関係に影響を与える要因以外でも特徴量を作ることが出来るため記述子という名前で総称しているのではと思います。)
Case1:元素数情報のみを使用
Case2:元素数情報、原子の合計数(nsites)、原子の種類数(nelements)使用
[IN]
atom_num_cols = ["Ac", "Ag", "Al", "Ar", "As", "Au", "B", "Ba", "Be", "Bi",
"Br", "C", "Ca", "Cd", "Ce", "Cl", "Co", "Cr", "Cs", "Cu", "Dy", "Er",
"Eu", "F", "Fe", "Ga", "Gd", "Ge", "H", "He", "Hf", "Hg", "Ho", "I",
"In", "Ir", "K", "Kr", "La", "Li", "Lu", "Mg", "Mn", "Mo", "N", "Na",
"Nb", "Nd", "Ne", "Ni", "Np", "O", "Os", "P", "Pa", "Pb", "Pd", "Pm",
"Pr", "Pt", "Pu", "Rb", "Re", "Rh", "Ru", "S", "Sb", "Sc", "Se", "Si",
"Sm", "Sn", "Sr", "Ta", "Tb", "Tc", "Te", "Th", "Ti", "Tl", "Tm", "U",
"V", "W", "Xe", "Y", "Yb", "Zn", "Zr"]
numeric_parameter_cols = atom_num_cols + ["nsites", "nelements"] #上記に原子の合計数と元素の種類数を追加
print(f'Case1: {len(atom_num_cols)}, Case2: {len(numeric_parameter_cols)}')
print(f'Case1: {atom_num_cols}')
print(f'Case2: {numeric_parameter_cols}')
[OUT]
Case1: 89, Case2: 91
Case1: ['Ac', 'Ag', 'Al', 'Ar', 'As', 'Au', 'B', 'Ba', 'Be', 'Bi', 'Br', 'C', 'Ca', 'Cd', 'Ce', 'Cl', 'Co', 'Cr', 'Cs', 'Cu', 'Dy', 'Er', 'Eu', 'F', 'Fe', 'Ga', 'Gd', 'Ge', 'H', 'He', 'Hf', 'Hg', 'Ho', 'I', 'In', 'Ir', 'K', 'Kr', 'La', 'Li', 'Lu', 'Mg', 'Mn', 'Mo', 'N', 'Na', 'Nb', 'Nd', 'Ne', 'Ni', 'Np', 'O', 'Os', 'P', 'Pa', 'Pb', 'Pd', 'Pm', 'Pr', 'Pt', 'Pu', 'Rb', 'Re', 'Rh', 'Ru', 'S', 'Sb', 'Sc', 'Se', 'Si', 'Sm', 'Sn', 'Sr', 'Ta', 'Tb', 'Tc', 'Te', 'Th', 'Ti', 'Tl', 'Tm', 'U', 'V', 'W', 'Xe', 'Y', 'Yb', 'Zn', 'Zr']
Case2: ['Ac', 'Ag', 'Al', 'Ar', 'As', 'Au', 'B', 'Ba', 'Be', 'Bi', 'Br', 'C', 'Ca', 'Cd', 'Ce', 'Cl', 'Co', 'Cr', 'Cs', 'Cu', 'Dy', 'Er', 'Eu', 'F', 'Fe', 'Ga', 'Gd', 'Ge', 'H', 'He', 'Hf', 'Hg', 'Ho', 'I', 'In', 'Ir', 'K', 'Kr', 'La', 'Li', 'Lu', 'Mg', 'Mn', 'Mo', 'N', 'Na', 'Nb', 'Nd', 'Ne', 'Ni', 'Np', 'O', 'Os', 'P', 'Pa', 'Pb', 'Pd', 'Pm', 'Pr', 'Pt', 'Pu', 'Rb', 'Re', 'Rh', 'Ru', 'S', 'Sb', 'Sc', 'Se', 'Si', 'Sm', 'Sn', 'Sr', 'Ta', 'Tb', 'Tc', 'Te', 'Th', 'Ti', 'Tl', 'Tm', 'U', 'V', 'W', 'Xe', 'Y', 'Yb', 'Zn', 'Zr', 'nsites', 'nelements']4-3.モデルの作成・学習
下記流れでモデル(LightGBM)を作成していきます。
交差検証の分割数(fold)を設定
LightGBMで使用するハイパーパラメータを設定
入力値(Pandasでカラムを指定)、ラベルを作成
ループ処理においてenumerate()のindexからfold番号を抽出できるよう、事前に4-1節で作成したfoldデータをNumpy型で用意しておく(val_fold)。
"val_fold=index番号"でmask(boolのリスト)を作成できるため、maskをデータに渡すことで学習用・検証用データに分割
モデルを作成・学習・推論を実施
各foldでモデルをPickleファイルで保存し、modelsリストに格納(次節でアンサンブルによる各モデルの平均値を推論値とします)
検証用データの性能(RMSE)を確認+各foldの平均値を算出
$$
評価指標:RSME=\sqrt{\frac{1}{N}\sum_{i=1}^{N}(y_i-\hat{y_i})^2}
$$
N:データ数、$${y_i}$$:正解値(ラベル)、$${\hat{y_i}}$$:推論値
Case1・2は入力値が変わるだけそれ以外はすべて同じです。Case1の方をコメントアウトした状態でコード記載しました。
なおLightGBMのearly_stoppingの設定詳細は下記の通りです。
[IN]
#交差検証Params
n_folds = 5 #分割数
#LightGBMのParams
lgbm_params = {
"objective": "regression", #回帰問題
"learning_rate": 0.2, #学習率
"reg_alpha": 0.1, #L1正則化
"reg_lambda": 0.1, #L2正則化
"random_state": 42, #乱数シード
"max_depth": 5, #木の深さ
"n_estimators": 1000000, #学習回数
"colsample_bytree": 0.9,
}
#LightGBMによるモデル作成:Case1
# X = df_train[atom_num_cols].values #説明変数
#LightGBMによるモデル作成:Case2
X = df_train[numeric_parameter_cols].values #説明変数
targets = df_train['formation_energy_per_atom'].values #目的変数
models = [] #モデルを格納するリスト
oof = np.zeros(df_train['formation_energy_per_atom'].shape[0]) #Out Of Foldの予測値を格納する配列
val_fold = df_train['val_fold'].values #foldの列を取得(0~4):Numpy配列に変換
for i_fols in tqdm(range(n_folds)):
print(f'Fold {i_fols + 1} / {n_folds}')
X_train, y_train = X[val_fold != i_fols], targets[val_fold != i_fols] #fold番号がi_folsでないデータを学習データとして取得
X_val, y_val = X[val_fold == i_fols], targets[val_fold == i_fols] #fold番号がi_folsのデータを検証データとして取得
#モデルの学習
model = lgbm.LGBMRegressor(**lgbm_params) #モデルの作成
model.fit(X_train, y_train,
eval_set = [(X_val, y_val)],
eval_metric='rmse',
callbacks=[lgbm.early_stopping(stopping_rounds=20, verbose=True),# early_stopping_rounds()の代わりにcallbacksを使う
lgbm.log_evaluation(400)]) #400回ごとに検証データのRMSEを表示
#モデルの予測
pred = model.predict(X_val)
#Out Of Foldの予測値を格納
oof[val_fold == i_fols] = model.predict(X_val) #fold番号がi_folsのデータの予測値を格納
cv_fold_rmse = np.sqrt(mean_squared_error(y_val, pred)) #fold番号がi_folsのデータのRMSEを計算
print(f'CV-fold {i_fols + 1} RMSE: {cv_fold_rmse:.4f}')
#各foldのモデルを保存
filename_model = f'model/Case1_fold{i_fols}.pkl' #モデルの保存先
with open(filename_model, 'wb') as f:
pickle.dump(model, f) #モデルの保存
models.append(model) #モデルをリストに追加
cv_rmse = np.sqrt(mean_squared_error(targets, oof)) #Out Of FoldのRMSEを計算
print(f'CV RMSE: {cv_rmse:.4f}')
[OUT]
0%| | 0/5 [00:00<?, ?it/s]Fold 1 / 5
Training until validation scores don't improve for 20 rounds
[400] valid_0's rmse: 0.433144 valid_0's l2: 0.187614
[800] valid_0's rmse: 0.399577 valid_0's l2: 0.159661
[1200] valid_0's rmse: 0.388458 valid_0's l2: 0.1509
[1600] valid_0's rmse: 0.380757 valid_0's l2: 0.144976
Early stopping, best iteration is:
[1894] valid_0's rmse: 0.37765 valid_0's l2: 0.142619
20%|██ | 1/5 [00:02<00:09, 2.26s/it]CV-fold 1 RMSE: 0.3776
Fold 2 / 5
Training until validation scores don't improve for 20 rounds
[400] valid_0's rmse: 0.419922 valid_0's l2: 0.176335
[800] valid_0's rmse: 0.394341 valid_0's l2: 0.155505
[1200] valid_0's rmse: 0.385389 valid_0's l2: 0.148525
[1600] valid_0's rmse: 0.379933 valid_0's l2: 0.144349
Early stopping, best iteration is:
[1920] valid_0's rmse: 0.376542 valid_0's l2: 0.141784
40%|████ | 2/5 [00:04<00:06, 2.24s/it]CV-fold 2 RMSE: 0.3765
Fold 3 / 5
Training until validation scores don't improve for 20 rounds
[400] valid_0's rmse: 0.412189 valid_0's l2: 0.1699
[800] valid_0's rmse: 0.38121 valid_0's l2: 0.145321
[1200] valid_0's rmse: 0.37132 valid_0's l2: 0.137879
Early stopping, best iteration is:
[1530] valid_0's rmse: 0.366708 valid_0's l2: 0.134475
60%|██████ | 3/5 [00:06<00:04, 2.06s/it]CV-fold 3 RMSE: 0.3667
Fold 4 / 5
Training until validation scores don't improve for 20 rounds
[400] valid_0's rmse: 0.431976 valid_0's l2: 0.186604
[800] valid_0's rmse: 0.405389 valid_0's l2: 0.164341
[1200] valid_0's rmse: 0.39489 valid_0's l2: 0.155938
[1600] valid_0's rmse: 0.389077 valid_0's l2: 0.151381
Early stopping, best iteration is:
[1951] valid_0's rmse: 0.385079 valid_0's l2: 0.148286
80%|████████ | 4/5 [00:08<00:02, 2.20s/it]CV-fold 4 RMSE: 0.3851
Fold 5 / 5
Training until validation scores don't improve for 20 rounds
[400] valid_0's rmse: 0.428916 valid_0's l2: 0.183969
[800] valid_0's rmse: 0.401205 valid_0's l2: 0.160966
Early stopping, best iteration is:
[1177] valid_0's rmse: 0.393401 valid_0's l2: 0.154764
CV-fold 5 RMSE: 0.3934
100%|██████████| 5/5 [00:10<00:00, 2.04s/it]
CV RMSE: 0.38004-4.特徴量の重要度を可視化
決定木はどのパラメータ(入力値)の影響が強いかを可視化することが出来ます(シンプルな決定木なら「dtreeviz」がオススメですが、GDBTには適用できないため普通の棒グラフを使用します)。
[IN]
plt.style.use('seaborn-whitegrid')
df_imp = pd.DataFrame(models[0].feature_importances_,
index = numeric_parameter_cols,
columns=['importance']).sort_values('importance', ascending=False)
plt.subplots(figsize=(len(numeric_parameter_cols) // 4, 5))
plt.bar(df_imp.index, df_imp.importance)
plt.grid()
plt.xticks(rotation=90)
plt.ylabel("importance")
plt.tight_layout()
plt.show()
[OUT]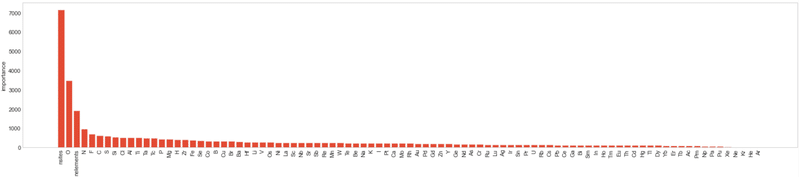
4-5.提出用(テスト)データの推論
学習したモデルを使用して提出用(テスト)データを作成します。フローは下記の通りです。
テスト用データを読み込み、入力値にするカラムだけ抽出してNumpy型に変更する。
各foldの学習済みモデルをmodelsに格納しているため、ループ処理で各modelで推論した後に値の平均値をとる※
(※今回は回帰(数値の予想)のため単純平均を使用しました。もしカテゴリカルデータなデータであれば多数決などで取ったりします。)
[IN]
#Case1
# X_test = df_test[atom_num_cols].values #テストデータの説明変数
#Case2
X_test = df_test[numeric_parameter_cols].values #テストデータの説明変数
preds = np.zeros(X_test.shape[0]) #テストデータの予測値を格納する配列
print(df_test.shape, preds.shape)
#アンサンブル学習:全モデルの結果を平均化
for model in models:
pred_fold = model.predict(X_test) #各foldのモデルでテストデータの予測値を計算
preds += pred_fold / n_folds #各foldの予測値を平均化
[OUT]
(28691, 94) (28691,)4-6.提出・結果確認
最後に指定の方式に変更してサイトから提出します。
(提出方式のCSVを読み込んで値だけ推論値に上書きしました)
[IN]
output = df_submit.copy() #提出用のデータフレームを作成
output['formation_energy_per_atom'] = preds #予測値を置換
# output.to_csv('output/submission_Case1.csv', index=False) #提出用のデータフレームをCSVファイルに出力
output.to_csv('output/submission_Case2.csv', index=False) #提出用のデータフレームをCSVファイルに出力
[OUT]参考までにCase1・2の学習時の平均RMSEはそれぞれ$${RMSE_{case1}=0.428}$$、$${RMSE_{case2}=0.380}$$のため、検証データとテストデータに大きな乖離が無いことが確認できました。


5.モデル実装2:CGCNN
5-1.CGCNNとは
XieとGrossmanによって提案された”Crystal Graph Convolutional neural network (CGCNN)”とは結晶グラフ(Crystal Graph)記述子を使用したモデルとのことです。
必要な情報は物質の結晶構造のみであり、結晶構造から結晶グラフ構造を作り、それをもとに機械学習モデルを作成します。重要なことは、記述子が物質構造の特徴を根底からうまく捉えられていないと、様々な物性に対応できる転移学習は行えません(つまりドメイン知識を元にした特徴量エンジニアリングが必要です)。
CGCNNは、結晶構造からグラフ構造をつくる部分と、埋め込み層、畳み込み層、プーリング層、全結合層からなる深層CNNの部分からなります。詳細は難しすぎて理解できなかったので、下記参考資料と文献をご確認ください。

【参考資料】
5-2.CIFの解析:Pymatgen
CIF(Crystallographic Information File: 結晶の構造情報)を解析できるライブラリとしてMaterials Projectより提供されているPymatgen (Python Materials Genomics)を使用していきます。
出力のイメージとして入力値にCIFを入れて、出力値で解析値を取得できます。また下図の通りグラフィックな出力も対応可能です。

まず初めにCIFを理解するためにデータNo.1のCIFを確認します。String型で結晶情報が記載されておりますがかなり見にくい状態です。これをPymatgenでParse(解析)していきます。
[IN]
cif_str = df_train['cif'][0]
print(type(cif_str), end='\n\n')
print(cif_str)
[OUT]
<class 'str'>
# generated using pymatgen
data_K2MgMo2(H2O5)2
_symmetry_space_group_name_H-M 'P 1'
_cell_length_a 6.00896235
_cell_length_b 6.55026940
_cell_length_c 7.95639355
_cell_angle_alpha 111.39242719
_cell_angle_beta 96.77130034
_cell_angle_gamma 109.07338782
_symmetry_Int_Tables_number 1
_chemical_formula_structural K2MgMo2(H2O5)2
_chemical_formula_sum 'K2 Mg1 Mo2 H4 O10'
_cell_volume 265.39072919
_cell_formula_units_Z 1
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
K K0 1 0.33066327 0.70285292 0.24630803 1
K K1 1 0.66933673 0.29714708 0.75369197 1
Mg Mg2 1 -0.00000000 -0.00000000 -0.00000000 1
Mo Mo3 1 0.33274891 0.66534980 0.76077634 1
Mo Mo4 1 0.66725109 0.33465020 0.23922366 1
H H5 1 0.92202881 0.86654805 0.28439073 1
H H6 1 0.07797119 0.13345195 0.71560927 1
H H7 1 0.12465733 0.12755969 0.37626561 1
H H8 1 0.87534267 0.87244031 0.62373439 1
O O9 1 0.36066563 0.82312200 0.62054963 1
O O10 1 0.63933437 0.17687800 0.37945037 1
O O11 1 0.23666938 0.34989125 0.61648940 1
O O12 1 0.76333062 0.65010875 0.38351060 1
O O13 1 0.62693462 0.76248819 0.92136504 1
O O14 1 0.37306538 0.23751181 0.07863496 1
O O15 1 0.10929347 0.70888614 0.89009389 1
O O16 1 0.89070653 0.29111386 0.10990611 1
O O17 1 0.05337462 0.97851338 0.25767835 1
O O18 1 0.94662538 0.02148662 0.74232165 15-2-1.結晶構造のParse:Structure
CIF(String型)からPymatgen.coreのStructureを使用してParseします。(printとdisplay方式で見え方が変わりますが)サマリー方式で結果を出力できました。
[IN]
from pymatgen.core import Structure
cif_str = df_train['cif'][0] #1つ目のcifを取得
crystal = Structure.from_str(cif_str, fmt='cif') #cifを構造体に変換
print(type(crystal))
print(crystal)
display(crystal)
[OUT]
<class 'pymatgen.core.structure.Structure'>
Structureには使用できる属性があり”specie”で原子名を抽出できます。
[IN]
for i in crystal:
print(i.specie.name, type(i.specie.name))
[OUT]
K <class 'str'>
K <class 'str'>
Mg <class 'str'>
Mo <class 'str'>
Mo <class 'str'>
H <class 'str'>
H <class 'str'>
H <class 'str'>
H <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>
O <class 'str'>5-2-2.近傍原子の情報抽出:get_all_neighbors
Structureのget_all_neighborsメソッドを使用するとradiusに指定した距離よりも近い各原子の近傍の原子のリストを取得することができます。
[get_all_neighbors]
get_all_neighbors(r: float, include_index: bool = False,
include_image: bool = False,
sites: Optional[Sequence[pymatgen.core.sites.PeriodicSite]] = None,
numerical_tol: float = 1e-08)→ list[list[pymatgen.core.structure.PeriodicNeighbor]]"get_all_neighbors"では各原子ごとに処理するため原子数(nsites)と同じ数のリストを出力します。
1つの原子に対して”各原子の近傍の原子のリスト”がリストとして入っており、100個以上のパターンが記載されています。
[IN]
nbrs = crystal.get_all_neighbors(r=8, include_index=True) #radiusに指定した距離内の原子を取得
atom_names = ['K','K','Mg','Mo','Mo','H','H','H','H','O','O','O','O','O','O','O','O','O','O']
print(type(nbrs), len(nbrs)) #nbrsの数=原子数(nsites)
for nbr, name in zip(nbrs, atom_names):
print(name, len(nbr), type(nbr))
nbrs[0][:5] #スライスで5つの近傍原子を取得
[OUT]
<class 'list'> 19
K 151 <class 'list'>
K 151 <class 'list'>
Mg 160 <class 'list'>
Mo 152 <class 'list'>
Mo 152 <class 'list'>
H 157 <class 'list'>
H 157 <class 'list'>
H 155 <class 'list'>
H 155 <class 'list'>
O 146 <class 'list'>
O 146 <class 'list'>
O 148 <class 'list'>
O 148 <class 'list'>
O 150 <class 'list'>
O 150 <class 'list'>
O 146 <class 'list'>
O 146 <class 'list'>
O 160 <class 'list'>
O 160 <class 'list'>
[PeriodicSite: O (-3.3835, -0.9887, -2.1435) [-0.6393, -0.1769, -0.3795],
PeriodicSite: Mo (-3.1652, -1.8707, -0.6311) [-0.6673, -0.3347, -0.2392],
PeriodicSite: O (-5.5961, -0.1201, 2.7722) [-0.9466, -0.0215, 0.2577],
PeriodicSite: K (-3.2692, -1.6610, 3.1439) [-0.6693, -0.2971, 0.2463],
PeriodicSite: O (-4.6048, -1.6273, 0.4521) [-0.8907, -0.2911, -0.1099]]なお"get_all_neighbors"の出力は”PeriodicNeighborクラス”であり、出力値は(site, distance, index, image)の順に出力されます。
[classPeriodicNeighbor]
classPeriodicNeighbor(species: pymatgen.core.composition.Composition,
coords: numpy.ndarray,
lattice: pymatgen.core.lattice.Lattice,
properties: Optional[dict] = None,
nn_distance: float = 0.0,
index: int = 0,
image: tuple = (0, 0, 0))[IN]
display(nbrs[0][0])
print(f'データ数:{len(nbrs[0][0])},データ型: {type(nbrs[0][0])}')
for idx, data in enumerate(nbrs[0][0]):
print(f'{idx}: {data}')
[OUT]
PeriodicSite: O (-3.3835, -0.9887, -2.1435) [-0.6393, -0.1769, -0.3795]
データ数:4,データ型: <class 'pymatgen.core.structure.PeriodicNeighbor'>
0: [-3.38 -0.99 -2.14] O
1: 6.4995036029880335
2: 9
3: (-1.0, -1.0, -1.0)5-3.結晶グラフ特徴量作成のシリアル実行用の関数
理解が難しいですが、おそらく次に”Joblib.Parallelによる並列計算”を実施するための関数を作成しています。中身は下記の通りです。
pymatgenでcif情報をparseして、”get_all_neighbors”メソッドで”各原子の近傍の原子のリスト”を取得
”各原子の近傍の原子のリスト””距離の近い順にソート
get_all_neighborsのindex=1で距離、2でindexを抽出していく。取得数(max_num_nbr)を指定して、CIF情報が指定数以下なら0埋め、指定数以上なら頭から指定数まで抽出してそれ以降は切り捨て
Structure.specieで原子名を抽出して{原子名:インデックス}の辞書で数値に変換
出力として原子数:nsitesと同じ数を持つデータ(特徴量とindexは取得数:max_num_nbr分だけ列方向に情報を持つ)
[IN]
atom_num_cols = ["Ac", "Ag", "Al", "Ar", "As", "Au", "B", "Ba", "Be", "Bi", "Br", "C", "Ca", "Cd", "Ce", "Cl", "Co", "Cr", "Cs", "Cu", "Dy", "Er",
"Eu", "F", "Fe", "Ga", "Gd", "Ge", "H", "He", "Hf", "Hg", "Ho", "I", "In", "Ir", "K", "Kr", "La", "Li", "Lu", "Mg", "Mn", "Mo", "N", "Na",
"Nb", "Nd", "Ne", "Ni", "Np", "O", "Os", "P", "Pa", "Pb", "Pd", "Pm", "Pr", "Pt", "Pu", "Rb", "Re", "Rh", "Ru", "S", "Sb", "Sc", "Se", "Si",
"Sm", "Sn", "Sr", "Ta", "Tb", "Tc", "Te", "Th", "Ti", "Tl", "Tm", "U", "V", "W", "Xe", "Y", "Yb", "Zn", "Zr"]
#原子のカテゴリ変数辞書: {原子名:インデックス}
atom_dict = {atom:idx for idx, atom in enumerate(atom_num_cols)}
print(f"原子の種類数:{len(atom_dict)}")
cif_str_list = df_train['cif'].to_numpy()
#結晶グラフ特徴量作成のシリアル実行
max_num_nbr = 12 #近傍原子の最大数
radius = 8 #近傍原子を探索する半径
features = [] #特徴量を格納するリスト
crystal = Structure.from_str(cif_str_list[0], fmt='cif') #cif文字列から構造を作成
all_nbrs = crystal.get_all_neighbors(r=radius, include_index=True) #8Å以内の原子のインデックスと距離を取得
all_nbrs = [sorted(nbrs, key=lambda x: x[1]) for nbrs in all_nbrs] #距離の近い順にソート
#近傍原子のインデックスと距離を格納するリスト
nbr_fea_idx = [] #get_all_neighborsのindex=2
nbr_fea = [] #get_all_neighborsのindex=1
for nbr in all_nbrs:
#近傍原子が足りない場合は0を格納
if len(nbr) < max_num_nbr:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr)) + [0] * (max_num_nbr - len(nbr))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr)) + [radius + 1.0] * (max_num_nbr - len(nbr))) #近傍原子の距離を格納
#近傍原子が多い場合は切り捨て(max_num_nbr個のみ格納)
else:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr[:max_num_nbr]))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr[:max_num_nbr]))) #近傍原子の距離を格納
nbr_fea_idx = np.array(nbr_fea_idx) #近傍原子のインデックスをnumpy配列に変換
nbr_fea = np.array(nbr_fea) #近傍原子の距離をnumpy配列に変換
atom_fea = [] #原子の特徴量を格納するリスト
for i in range(len(crystal)):
atom_fea.append(atom_dict[crystal[i].specie.name]) #原子の特徴量を格納
atom_fea = np.array(atom_fea) #原子の特徴量をnumpy配列に変換
print(f'atom_fea形状:{atom_fea.shape}')
print(f'atom_fea:{atom_fea}')
print(f'nbr_fea_idx形状:{nbr_fea_idx.shape}')
print(f'nbr_fea_idx:{nbr_fea_idx}')
print(f'nbr_fea形状:{nbr_fea.shape}')
print(f'nbr_fea:{nbr_fea}')
[OUT]
原子の種類数:89
atom_fea形状:(19,)
atom_fea:[36 36 41 43 43 28 28 28 28 51 51 51 51 51 51 51 51 51 51]
nbr_fea_idx形状:(19, 12)
nbr_fea_idx:[[10 9 17 16 12 14 5 15 7 5 13 4]
[ 9 10 18 15 11 13 6 16 8 6 14 3]
[18 17 13 14 15 16 8 7 5 6 4 3]
[ 9 11 13 15 6 7 8 14 1 2 8 18]
[10 12 14 16 5 8 7 13 0 2 7 17]
[17 7 12 8 2 13 0 10 11 6 4 0]
[18 8 11 7 2 14 1 9 12 5 3 1]
[17 5 11 2 6 10 10 16 14 12 8 0]
[18 6 12 2 5 9 9 15 13 11 7 1]
[ 3 1 0 11 15 13 8 6 8 18 18 12]
[ 4 0 1 12 16 14 7 5 7 17 17 11]
[ 7 3 6 17 18 1 13 9 15 5 8 10]
[ 8 4 5 18 17 0 14 10 16 6 7 9]
[ 3 2 11 5 9 1 16 18 15 17 15 8]
[ 4 2 12 6 10 0 15 17 16 18 16 7]
[ 3 2 1 9 17 11 14 13 0 18 13 8]
[ 4 2 0 10 18 12 13 14 1 17 14 7]
[ 5 7 2 11 0 12 15 14 13 16 10 10]
[ 6 8 2 12 1 11 16 13 14 15 9 9]]
nbr_fea形状:(19, 12)
nbr_fea:[[2.75 2.75 2.82 2.82 2.87 2.96 2.99 3.01 3.29 3.3 3.37 3.61]
[2.75 2.75 2.82 2.82 2.87 2.96 2.99 3.01 3.29 3.3 3.37 3.61]
[2.1 2.1 2.13 2.13 2.14 2.14 2.73 2.73 2.74 2.74 3.63 3.63]
[1.76 1.8 1.82 1.82 3.18 3.42 3.56 3.58 3.61 3.63 3.66 3.72]
[1.76 1.8 1.82 1.82 3.18 3.42 3.56 3.58 3.61 3.63 3.66 3.72]
[0.99 1.57 1.91 2.73 2.74 2.92 2.99 3. 3.06 3.08 3.18 3.3 ]
[0.99 1.57 1.91 2.73 2.74 2.92 2.99 3. 3.06 3.08 3.18 3.3 ]
[0.99 1.57 1.8 2.73 2.73 3. 3.04 3.09 3.12 3.2 3.22 3.29]
[0.99 1.57 1.8 2.73 2.73 3. 3.04 3.09 3.12 3.2 3.22 3.29]
[1.76 2.75 2.75 2.93 2.94 2.94 3. 3. 3.04 3.21 3.25 3.45]
[1.76 2.75 2.75 2.93 2.94 2.94 3. 3. 3.04 3.21 3.25 3.45]
[1.8 1.8 1.91 2.78 2.86 2.87 2.9 2.93 2.95 3.06 3.2 3.45]
[1.8 1.8 1.91 2.78 2.86 2.87 2.9 2.93 2.95 3.06 3.2 3.45]
[1.82 2.13 2.9 2.92 2.94 2.96 2.97 2.98 2.98 3. 3.06 3.12]
[1.82 2.13 2.9 2.92 2.94 2.96 2.97 2.98 2.98 3. 3.06 3.12]
[1.82 2.14 2.82 2.94 2.94 2.95 2.97 2.98 3.01 3.05 3.06 3.09]
[1.82 2.14 2.82 2.94 2.94 2.95 2.97 2.98 3.01 3.05 3.06 3.09]
[0.99 0.99 2.1 2.78 2.82 2.86 2.94 2.98 3. 3.05 3.21 3.25]
[0.99 0.99 2.1 2.78 2.82 2.86 2.94 2.98 3. 3.05 3.21 3.25]]
上記の処理を関数化します。
[IN]
#結晶グラフ特徴量作成のシリアル実行
def _graph_features(cif_str, idx, atom_dict, max_num_nbr=12, radius=8):
features = [] #特徴量を格納するリスト
crystal = Structure.from_str(cif_str, fmt='cif') #cif文字列から構造を作成
all_nbrs = crystal.get_all_neighbors(r=radius, include_index=True) #8Å以内の原子のインデックスと距離を取得
all_nbrs = [sorted(nbrs, key=lambda x: x[1]) for nbrs in all_nbrs] #距離の近い順にソート
#近傍原子のインデックスと距離を格納するリスト
nbr_fea_idx = [] #get_all_neighborsのindex=2
nbr_fea = [] #get_all_neighborsのindex=1
for nbr in all_nbrs:
#近傍原子が足りない場合は0を格納
if len(nbr) < max_num_nbr:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr)) + [0] * (max_num_nbr - len(nbr))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr)) + [radius + 1.0] * (max_num_nbr - len(nbr))) #近傍原子の距離を格納
#近傍原子が多い場合は切り捨て(max_num_nbr個のみ格納)
else:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr[:max_num_nbr]))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr[:max_num_nbr]))) #近傍原子の距離を格納
nbr_fea_idx = np.array(nbr_fea_idx) #近傍原子のインデックスをnumpy配列に変換
nbr_fea = np.array(nbr_fea) #近傍原子の距離をnumpy配列に変換
atom_fea = [] #原子の特徴量を格納するリスト
for i in range(len(crystal)):
atom_fea.append(atom_dict[crystal[i].specie.name]) #原子の特徴量を格納
atom_fea = np.array(atom_fea) #原子の特徴量をnumpy配列に変換
return (atom_fea, nbr_fea_idx, nbr_fea), idx
[OUT]5-4.並列処理+特徴量をdfに追加
本節では処理を高速化(並列処理)するためにjoblib.Parallelを使用します。
[class joblib.Parallel]
joblib.Parallel(n_jobs=None, backend=None, verbose=0,
timeout=None, pre_dispatch='2 * n_jobs',
batch_size='auto', temp_folder=None, max_nbytes='1M',
mmap_mode='r', prefer=None, require=None) 実際に処理をすると全データ(訓練データ:71729)処理して時間がかかるため2個だけ抽出しました。結果として"[<関数の出力1>, <関数の出力2>・・]"のような形で出力され、先ほど作成した"graph_feature"関数を渡すので"(atom_fea, nbr_fea_idx, nbr_fea), idx"が出力されます。
なお渡す値はDataFrameからNumpy(Series.to_numpy())に変更しないと処理が死ぬほど遅くなります。
[IN]
processed = joblib.Parallel(n_jobs=-1, verbose=1)(joblib.delayed(_graph_feature)(cif_str, idx, atom_dict) for idx, cif_str in enumerate(cif_str_list[:2]))
print(len(processed), type(processed))
print(len(processed[0]), type(processed[0]))
print(f'feats_tup: {len(processed[0][0])}, index: {processed[0][1]}')
[OUT]
2 <class 'list'>
2 <class 'tuple'>
feats_tup: 3, index: 0前節で作成した特徴量抽出関数でデータを処理して、その結果(site、距離、index)をdfに追加します。なお本処理は時間がかかるため1回処理したら次回以降は処理後のファイルを読み込めるように”numpy.savez"で配列データを保存しました。
参考として処理後のDataFrameから5つ抜き出して結果を表示しました。
[IN]
df_train = pd.read_csv(file_train) #訓練用データ(train.csv)
atom_num_cols = ["Ac", "Ag", "Al", "Ar", "As", "Au", "B", "Ba", "Be", "Bi", "Br", "C", "Ca", "Cd", "Ce", "Cl", "Co", "Cr", "Cs", "Cu", "Dy", "Er",
"Eu", "F", "Fe", "Ga", "Gd", "Ge", "H", "He", "Hf", "Hg", "Ho", "I", "In", "Ir", "K", "Kr", "La", "Li", "Lu", "Mg", "Mn", "Mo", "N", "Na",
"Nb", "Nd", "Ne", "Ni", "Np", "O", "Os", "P", "Pa", "Pb", "Pd", "Pm", "Pr", "Pt", "Pu", "Rb", "Re", "Rh", "Ru", "S", "Sb", "Sc", "Se", "Si",
"Sm", "Sn", "Sr", "Ta", "Tb", "Tc", "Te", "Th", "Ti", "Tl", "Tm", "U", "V", "W", "Xe", "Y", "Yb", "Zn", "Zr"]
#原子のカテゴリ変数辞書: {原子名:インデックス}
atom_dict = {atom:idx for idx, atom in enumerate(atom_num_cols)}
print(f"原子の種類数:{len(atom_dict)}")
cif_str_list = df_train['cif'].to_numpy()
#結晶グラフ特徴量作成のシリアル実行
def _graph_features(cif_str, idx, atom_dict, max_num_nbr=12, radius=8):
features = [] #特徴量を格納するリスト
crystal = Structure.from_str(cif_str, fmt='cif') #cif文字列から構造を作成
all_nbrs = crystal.get_all_neighbors(r=radius, include_index=True) #8Å以内の原子のインデックスと距離を取得
all_nbrs = [sorted(nbrs, key=lambda x: x[1]) for nbrs in all_nbrs] #距離の近い順にソート
#近傍原子のインデックスと距離を格納するリスト
nbr_fea_idx = [] #get_all_neighborsのindex=2
nbr_fea = [] #get_all_neighborsのindex=1
for nbr in all_nbrs:
#近傍原子が足りない場合は0を格納
if len(nbr) < max_num_nbr:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr)) + [0] * (max_num_nbr - len(nbr))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr)) + [radius + 1.0] * (max_num_nbr - len(nbr))) #近傍原子の距離を格納
#近傍原子が多い場合は切り捨て(max_num_nbr個のみ格納)
else:
nbr_fea_idx.append(list(map(lambda x: x[2], nbr[:max_num_nbr]))) #近傍原子のインデックスを格納
nbr_fea.append(list(map(lambda x: x[1], nbr[:max_num_nbr]))) #近傍原子の距離を格納
nbr_fea_idx = np.array(nbr_fea_idx) #近傍原子のインデックスをnumpy配列に変換
nbr_fea = np.array(nbr_fea) #近傍原子の距離をnumpy配列に変換
atom_fea = [] #原子の特徴量を格納するリスト
for i in range(len(crystal)):
atom_fea.append(atom_dict[crystal[i].specie.name]) #原子の特徴量を格納
atom_fea = np.array(atom_fea) #原子の特徴量をnumpy配列に変換
output = (atom_fea, nbr_fea_idx, nbr_fea) #結果をタプルに格納
return output, idx
#Joblibを使って並列処理
def graph_features(df, atom_dict, filename, max_num_nbr=12, radius=8):
cif_str_list = df['cif'].to_numpy() #cif文字列をリストに格納
processed = joblib.Parallel(n_jobs=-1, verbose=1)(joblib.delayed(_graph_features)(cif_str, idx, atom_dict) for idx, cif_str in enumerate(cif_str_list))
processed.sort(key=lambda x: x[1]) #実行されて帰ってきた結果は必ずしも、元データの順に並んでいないことがある
features_list = [fea[0] for fea in processed] #結果をリストに格納
#出力は(特徴量(tuple), index)のため特徴量だけ抽出
_features = [t[0] for t in processed]
nbr_fea_idx_list = [] #近傍原子(Neighbor)のindex
nbr_fea_list = [] #近傍原子の距離
atom_fea_list = [] #原子の種類
for fea in _features:
atom_fea_list.append(fea[0])
nbr_fea_idx_list.append(fea[1])
nbr_fea_list.append(fea[2])
#DataFrameに追加
df['atom_fea'] = atom_fea_list
df['nbr_fea'] = nbr_fea_list
df['nbr_fea_idx'] = nbr_fea_idx_list
#numpy配列で保存
filename = 'train_feats.npz'
filepath_feat = os.path.join('feats', filename)
np.savez(filepath_feat,
atom_fea=np.array(atom_fea_list),
nbr_fea_idx=np.array(nbr_fea_idx_list),
nbr_fea=np.array(nbr_fea_list))
return df
_df = df_train.head(5)
filename = 'train_feats(test).npz'
_dfwithfeat = graph_features(_df, atom_dict, filename)
_dfwithfeat
[OUT]
子の種類数:89
(5, 98)
※注記:上記だと”SettingWithCopyWarning”と”VisibleDeprecationWarning:”が消えないのですが、とりあえず欲しい結果が得られているためそのまま行きます。
5-5.ガウシアン距離の計算
(もうついていけなくなってますが)Gaussian basisを元に距離[Å]を拡張する関数を作成して、距離情報(nbr_fea)を再計算しています。
理解できないのでとりあえず結果だけ記載しておきます。もともと1個のデータは距離情報が12次元だったのが(12, 41)次元※に拡張されています(np.arange()でdmin,dmaxをstepで区切るため$${\frac{8-0}{0.2}+1}$$)。
[IN]
#Gaussian basis(ガウス基底関数)による距離[Å]特徴量の作成
class GaussianDistance(object):
def __init__(self, dmin, dmax, step, var=None):
assert dmin < dmax #最小原子間距離(dmin)が最大原子間距離(dmax)より小さいこと
assert dmax - dmin > step #最大原子間距離(dmax)と最小原子間距離(dmin)の差がガウシアンフィルタのステップ数(step)より大きいこと
self.filter = np.arange(dmin, dmax+step, step) #距離の範囲を指定
if var is None:
var = step
self.var = var #ガウシアンフィルタの分散(variance)
def expand(self, distance):
#ガウシアン距離フィルタを適用
output = np.exp(-(distance[..., np.newaxis] - self.filter)**2 / self.var**2)
return output
#1個のデータで確認
_fea = _dfwithfeat['nbr_fea'].to_numpy()[0]
print(_fea.shape, type(_fea)) #入力値(距離)の形状を確認
print(_fea, end='\n\n')
#距離をガウス分布に変換
_gauss = GaussianDistance(dmin=0.0, dmax=8.0, step=0.2)
_distance = _gauss.expand(_fea)
print(_distance.shape, type(_distance))
_distance[0]
[OUT]
(19, 12) <class 'numpy.ndarray'>
[[2.75 2.75 2.82 2.82 2.87 2.96 2.99 3.01 3.29 3.3 3.37 3.61]
[2.75 2.75 2.82 2.82 2.87 2.96 2.99 3.01 3.29 3.3 3.37 3.61]
[2.1 2.1 2.13 2.13 2.14 2.14 2.73 2.73 2.74 2.74 3.63 3.63]
[1.76 1.8 1.82 1.82 3.18 3.42 3.56 3.58 3.61 3.63 3.66 3.72]
[1.76 1.8 1.82 1.82 3.18 3.42 3.56 3.58 3.61 3.63 3.66 3.72]
[0.99 1.57 1.91 2.73 2.74 2.92 2.99 3. 3.06 3.08 3.18 3.3 ]
[0.99 1.57 1.91 2.73 2.74 2.92 2.99 3. 3.06 3.08 3.18 3.3 ]
[0.99 1.57 1.8 2.73 2.73 3. 3.04 3.09 3.12 3.2 3.22 3.29]
[0.99 1.57 1.8 2.73 2.73 3. 3.04 3.09 3.12 3.2 3.22 3.29]
[1.76 2.75 2.75 2.93 2.94 2.94 3. 3. 3.04 3.21 3.25 3.45]
[1.76 2.75 2.75 2.93 2.94 2.94 3. 3. 3.04 3.21 3.25 3.45]
[1.8 1.8 1.91 2.78 2.86 2.87 2.9 2.93 2.95 3.06 3.2 3.45]
[1.8 1.8 1.91 2.78 2.86 2.87 2.9 2.93 2.95 3.06 3.2 3.45]
[1.82 2.13 2.9 2.92 2.94 2.96 2.97 2.98 2.98 3. 3.06 3.12]
[1.82 2.13 2.9 2.92 2.94 2.96 2.97 2.98 2.98 3. 3.06 3.12]
[1.82 2.14 2.82 2.94 2.94 2.95 2.97 2.98 3.01 3.05 3.06 3.09]
[1.82 2.14 2.82 2.94 2.94 2.95 2.97 2.98 3.01 3.05 3.06 3.09]
[0.99 0.99 2.1 2.78 2.82 2.86 2.94 2.98 3. 3.05 3.21 3.25]
[0.99 0.99 2.1 2.78 2.82 2.86 2.94 2.98 3. 3.05 3.21 3.25]]
(19, 12, 41) <class 'numpy.ndarray'>
array([[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.04, 0.56, 0.95, 0.22, 0.01, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.04, 0.55, 0.95, 0.22, 0.01, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.01, 0.31, 0.99, 0.43, 0.02, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.01, 0.29, 0.99, 0.46, 0.03, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.15, 0.87, 0.67, 0.07, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.04, 0.53, 0.96, 0.23, 0.01, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.02, 0.41, 1. , 0.33, 0.01, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.02, 0.35, 1. , 0.39, 0.02, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.13, 0.83, 0.72, 0.08, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.11, 0.78, 0.78, 0.1 , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.03, 0.48, 0.98, 0.27, 0.01, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ],
[0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0.02, 0.33, 1. , 0.4 , 0.02, 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. , 0. ]])5-6.CIFの自作Dataset作成
先ほど特徴量を追加したDataFrameからDatasetを作成できるクラスを自作します。PytorchではDatasetクラスを継承して自作Datasetの作成が可能です。
内容としてはDataFrameを入力してCGCNNに必要な条件(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])をDatasetとして出力できるようにします。
[IN]
class CIFDataset(Dataset):
def __init__(self, dmin=0, step=0.2, max_num_nbr = 12, radius = 8, df = None, has_labels = False):
self.max_num_nbr = max_num_nbr #近傍原子の最大取得数
self.radius = radius #近傍原子の検索半径
self.len = len(df) #データ数
self.ids = df["id"].to_numpy() #データNo.(id)
self.nbr_fea_idx_arr = df["nbr_fea_idx"].to_numpy() #近傍原子のインデックス
self.nbr_fea_arr = df["nbr_fea"].to_numpy() #近傍原子の距離
self.atom_fea_arr = df["atom_fea"].to_numpy() #原子の特徴量
self.has_labels = has_labels
#ガウス分布による距離特徴量の作成
self.gdf = GaussianDistance(dmin=dmin, dmax=self.radius, step=step)
if self.has_labels:
self.targets = df['formation_energy_per_atom'].to_numpy() #目的変数
def __len__(self):
return self.len
def __getitem__(self, idx):
items = dict()
items["id"] = self.ids[idx]
if self.has_labels:
items["target"] = torch.as_tensor(self.targets[idx], dtype = torch.float32) #目的変数
items["atom_fea"] = torch.as_tensor(self.atom_fea_arr[idx], dtype = torch.int) #原子の特徴量
nbr_fea = self.gdf.expand(self.nbr_fea_arr[idx]) #近傍原子の距離をガウス分布に変換
items["nbr_fea"] = torch.as_tensor(nbr_fea, dtype = torch.float32) #近傍原子の距離
items["nbr_fea_idx"] = torch.as_tensor(self.nbr_fea_idx_arr[idx]) #近傍原子のインデックス
return items
[OUT]
-サンプルで確認すると入力したデータ数および必要なカラムデータが取得できること確認しました。
[IN]
_df = df_train.head(5)
filename = 'train_feats(test).npz'
_df = graph_feature(_df, atom_dict, filename)
dataset = CIFDataset(df = _df, has_labels = True)
for i in dataset:
print(type(i), i.keys())
print(len(dataset))
[OUT]
<class 'dict'> dict_keys(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])
<class 'dict'> dict_keys(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])
<class 'dict'> dict_keys(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])
<class 'dict'> dict_keys(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])
<class 'dict'> dict_keys(['id', 'target', 'atom_fea', 'nbr_fea', 'nbr_fea_idx'])
55-7.バッチ単位に集約
複数の結晶を1つのbatchにまとめます。結晶ごとの原子と近傍原子の組の情報を正しく設定するために、nbr_fea_idx(近傍原子の組のindex)は集約する際に更新し、crystal_atom_idx(各結晶の原子のindex)の情報を付与しています。
[IN]
from typing import List
#Listを照合して結晶構造のデータのバッチを作成
class CollateFn:
def __init__(self, has_labels = False):
self.has_labels = has_labels
def __call__(self, dataset: List):
batch_atom_fea, batch_nbr_fea, batch_nbr_fea_idx = [], [], []
crystal_atom_idx, batch_target = [], []
batch_ids = []
base_idx = 0
for i, item in enumerate(dataset):
n_i = item["atom_fea"].shape[0] #結晶構造の原子数
#バッチの作成(結晶構造のデータを追加)
batch_atom_fea.append(item["atom_fea"])
batch_nbr_fea.append(item["nbr_fea"])
batch_nbr_fea_idx.append(item["nbr_fea_idx"] + base_idx)
new_idx = torch.LongTensor(np.arange(n_i) + base_idx) #結晶構造の原子のインデックス
crystal_atom_idx.append(new_idx)
batch_ids.append(item["id"])
base_idx += n_i
#出力用の辞書を作成
output = dict()
output["atom_fea"] = torch.cat(batch_atom_fea, dim=0)
output["nbr_fea"] = torch.cat(batch_nbr_fea, dim=0)
output["nbr_fea_idx"] = torch.cat(batch_nbr_fea_idx, dim=0)
output["crystal_atom_idx"] = crystal_atom_idx
output["ids"] = batch_ids
if self.has_labels:
batch_target.append(item["target"])
output["targets"] = torch.stack(batch_target, dim=0)
return output
[OUT]ミニバッチサイズ=5のDatasetで動作確認してみます。出力は6つのKeyを持つ辞書であり、ラベル(生成エネルギー)などはバッチサイズと同じですが特徴量はひとまとめで作成されております。
[IN]
_df = df_train.head(5)
filename = 'train_feats(test).npz'
_df = graph_feature(_df, atom_dict, filename)
dataset = CIFDataset(df = _df, has_labels = True)
cf = CollateFn(has_labels = True)
batch = cf(dataset)
print(len(batch), batch.keys())
for key, value in batch.items():
if type(value) == list:
print(key, type(value), len(value))
else:
print(key, type(value), value.shape)
[OUT]
6 dict_keys(['atom_fea', 'nbr_fea', 'nbr_fea_idx', 'crystal_atom_idx', 'ids', 'targets'])
atom_fea <class 'torch.Tensor'> torch.Size([93])
nbr_fea <class 'torch.Tensor'> torch.Size([93, 12, 41])
nbr_fea_idx <class 'torch.Tensor'> torch.Size([93, 12])
crystal_atom_idx <class 'list'> 5
ids <class 'list'> 5
targets <class 'torch.Tensor'> torch.Size([5])5-8.訓練データの前処理
既に作成した”graph_features”を使用してCIFの結晶構造を解析していきます。並列計算処理を入れても時間がかかる(4.5min程度)ため2回目以降は保存したNumpyファイル(npz)を読み込むようにしました。
[IN]
feats_filename = "train_feats.npz"
feats_filepath = os.path.join(CFG.feats_dir, feats_filename)
keys = ["atom_fea", "nbr_fea", "nbr_fea_idx"]
if os.path.exists(feats_filepath):
graph_feats = np.load(feats_filepath, allow_pickle=True) #ファイルがあるならnpzファイルを読み込む
for k in keys:
train_df[k] = graph_feats[k] #train_dfに追加
else:
train_df = graph_features(train_df, atom_dict, feats_filename, max_num_nbr = CFG.max_num_nbr, radius = CFG.radius)
[OUT]
[Parallel(n_jobs=-1)]: Using backend LokyBackend with 16 concurrent workers.
[Parallel(n_jobs=-1)]: Done 18 tasks | elapsed: 0.1s
[Parallel(n_jobs=-1)]: Done 320 tasks | elapsed: 1.8s
[Parallel(n_jobs=-1)]: Done 2224 tasks | elapsed: 8.5s
[Parallel(n_jobs=-1)]: Done 5024 tasks | elapsed: 18.8s
[Parallel(n_jobs=-1)]: Done 8624 tasks | elapsed: 31.6s
[Parallel(n_jobs=-1)]: Done 13024 tasks | elapsed: 49.2s
[Parallel(n_jobs=-1)]: Done 18224 tasks | elapsed: 1.1min
[Parallel(n_jobs=-1)]: Done 24224 tasks | elapsed: 1.5min
[Parallel(n_jobs=-1)]: Done 31024 tasks | elapsed: 2.0min
[Parallel(n_jobs=-1)]: Done 38624 tasks | elapsed: 2.4min
[Parallel(n_jobs=-1)]: Done 47024 tasks | elapsed: 2.9min
[Parallel(n_jobs=-1)]: Done 56224 tasks | elapsed: 3.5min
[Parallel(n_jobs=-1)]: Done 66224 tasks | elapsed: 4.1min
[Parallel(n_jobs=-1)]: Done 71729 out of 71729 | elapsed: 4.5min finished5-9.CGCNNの実装
公開されているCrystal Graph Convolutional Neural Networks(CGCNN)の実装に倣い、ノードを組成式を構成する原子、エッジを近傍する原子の組として結晶グラフを作成します。
エッジ特徴量:radiusが8以内の近傍の原子の組の最大数を12としてその原子のindexとエッジの特徴量(結合している原子の距離)を取得しガウシアンフィルタ処理で41次元に変換した値。具体的にはpymatgen.core.IStructureのget_all_neighborsメソッドでr(radius of sphere)は8になっているのでそれに合わせて近傍の原子を取得
ノード特徴量:89種類の原子番号のカテゴリ変数の値を用意します。Entity Embeddingを行いカテゴリ変数の分散表現を学習する構造にしています。
※公開されているCGCNNの実装(data.py L314)ではatom_init.jsonに、原子番号ごとに固定値のベクトルが用意されております。
実装方針について公開されているCGCNNの実装と異なる点は、原子の特徴量は固定のベクトルではなく、学習可能なカテゴリ変数の分散表現としている点です。
(ちなみに名前を参照してConvLayerとしていますがただの全結合???Conv2dの畳み込みは今回入っておりません)
atom_nunique: 原子の種類数
nbr_fea_len: エッジ(近傍原子の組)の特徴量の次元 # nbr_fea_len = (dmax+step - dmin) / step = (8 + 0.2 - 0) / 0.2 = 41
atom_fea_len: embedding層で取得する次元の原子の分散表現
n_conv: 指定した回数だけの隠れ層の繰り返し、2 * atom_fea_len + nbr_fea_len次元の特徴量を入出力としてグラフ表現を学習する構造になっています
h_fea_len: 隠れ層での特徴量の次元
n_h: pooling層の後の隠れ層の数
[IN]
class ConvLayer(nn.Module):
def __init__(self, atom_fea_len, nbr_fea_len):
super(ConvLayer, self).__init__()
self.atom_fea_len = atom_fea_len #embedding層で取得する次元の原子の分散表現
self.nbr_fea_len = nbr_fea_len #エッジ(近傍原子の組)の特徴量の次元 # nbr_fea_len = (dmax+step - dmin) / step = (8 + 0.2 - 0) / 0.2 = 41
self.fc_full = nn.Linear(2*self.atom_fea_len + self.nbr_fea_len,
2*self.atom_fea_len) #全結合層
self.sigmoid = nn.Sigmoid() #シグモイド関数
self.softplus1 = nn.Softplus() #ソフトプラス関数
self.bn1 = nn.BatchNorm1d(2*self.atom_fea_len) #バッチ正規化
self.bn2 = nn.BatchNorm1d(self.atom_fea_len) #バッチ正規化
self.softplus2 = nn.Softplus() #ソフトプラス関数
def forward(self, atom_in_fea, nbr_fea, nbr_fea_idx):
N = nbr_fea_idx.shape[0] #バッチ内の総原子数
M = nbr_fea_idx.shape[1] #近傍原子の組の数
atom_nbr_fea = atom_in_fea[nbr_fea_idx, :] #近傍原子の特徴量
total_nbr_fea = torch.cat([atom_in_fea.unsqueeze(1).expand(N, M, self.atom_fea_len),
atom_nbr_fea, nbr_fea], dim=2) #結合原子の特徴量と近傍原子の特徴量を結合
y = self.fc_full(total_nbr_fea) #全結合層
y = self.bn1(y.view(-1, 2*self.atom_fea_len)).view(N, M, 2*self.atom_fea_len) #バッチ正規化
nbr_filter, nbr_core = y.chunk(2, dim=2) #チャンク分割
nbr_filter = self.sigmoid(nbr_filter) #シグモイド関数
nbr_core = self.softplus1(nbr_core) #ソフトプラス関数
nbr_sumed = torch.sum(nbr_filter * nbr_core, dim=1) #近傍原子の特徴量の加重和
nbr_sumed = self.bn2(nbr_sumed) #バッチ正規化
out = self.softplus2(atom_in_fea + nbr_sumed) #ソフトプラス関数
return out
class CrystalGraphConvNet(nn.Module):
def __init__(self, atom_nunique = 89, nbr_fea_len = 41, atom_fea_len = 64,
n_conv = 3, h_fea_len = 128, n_h = 1, classification = False):
super(CrystalGraphConvNet, self).__init__()
self.classification = classification
self.embedding = nn.Embedding(atom_nunique, atom_fea_len, padding_idx=0) #原子の分散表現
self.convs = nn.ModuleList([ConvLayer(atom_fea_len, nbr_fea_len) for _ in range(n_conv)])
self.conv_to_fc = nn.Linear(atom_fea_len, h_fea_len) #全結合層
self.conv_to_fc_softplus = nn.Softplus() #ソフトプラス関数
if n_h > 1:
self.fcs = nn.ModuleList([nn.Linear(h_fea_len, h_fea_len) for _ in range(n_h-1)]) #全結合層
self.softpluses = nn.ModuleList([nn.Softplus() for _ in range(n_h-1)]) #ソフトプラス関数
if self.classification:
self.fc_out = nn.Linear(h_fea_len, 2) #全結合層(入力層:128, 出力層:2)
else:
self.fc_out = nn.Linear(h_fea_len, 1) #全結合層(入力層:128, 出力層:1)
if self.classification:
self.logsoftmax = nn.LogSoftmax(dim=1) #対数ソフトマックス関数
self.dropout = nn.Dropout()
def forward(self, atom_fea, nbr_fea, nbr_fea_idx, crystal_atom_idx):
atom_fea = self.embedding(atom_fea) #原子由来の特徴量であり変化する値
for conv_func in self.convs:
atom_fea = conv_func(atom_fea, nbr_fea, nbr_fea_idx)
y = self.pooling(atom_fea, crystal_atom_idx) #原子ごとの特徴量を結合して結晶ごとの特徴量を作成
y = self.conv_to_fc(self.conv_to_fc_softplus(y)) #全結合層
y = self.conv_to_fc_softplus(y) #ソフトプラス関数
if self.classification:
y = self.dropout(y) #ドロップアウト
if hasattr(self, 'fcs') and hasattr(self, 'softpluses'):
for fc, softplus in zip(self.fcs, self.softpluses):
y = fc(y)
y = softplus(y)
out = self.fc_out(y) #全結合層
if self.classification:
out = self.logsoftmax(out)
return out
def pooling(self, atom_fea, crystal_atom_idx):
assert sum([len(x) for x in crystal_atom_idx]) == atom_fea.data.shape[0]
#バッチ内の結晶す鵜の合計(N0)のマッピング(crystal_atom_idx)から平均値を計算
summed_fea = [torch.mean(atom_fea[i], dim=0, keepdim=True) for i in crystal_atom_idx]
return torch.cat(summed_fea, dim=0)
[OUT]出力結果をサンプルで見てみます。
[IN]
item = dataset.__getitem__(2)
model = CrystalGraphConvNet(
atom_nunique = 89,
nbr_fea_len= item['nbr_fea'].shape[-1],
atom_fea_len= 64, #原子の分散表現の次元数
n_conv = 3,#Conv-Layerの数
h_fea_len= 64, #全結合層の出力数
n_h = 1, #全結合層の数
classification= False
)
model
[OUT]
CrystalGraphConvNet(
(embedding): Embedding(89, 64, padding_idx=0)
(convs): ModuleList(
(0): ConvLayer(
(fc_full): Linear(in_features=169, out_features=128, bias=True)
(sigmoid): Sigmoid()
(softplus1): Softplus(beta=1, threshold=20)
(bn1): BatchNorm1d(128, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(bn2): BatchNorm1d(64, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(softplus2): Softplus(beta=1, threshold=20)
)
(1): ConvLayer(
(fc_full): Linear(in_features=169, out_features=128, bias=True)
(sigmoid): Sigmoid()
(softplus1): Softplus(beta=1, threshold=20)
(bn1): BatchNorm1d(128, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(bn2): BatchNorm1d(64, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(softplus2): Softplus(beta=1, threshold=20)
)
(2): ConvLayer(
(fc_full): Linear(in_features=169, out_features=128, bias=True)
(sigmoid): Sigmoid()
(softplus1): Softplus(beta=1, threshold=20)
(bn1): BatchNorm1d(128, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(bn2): BatchNorm1d(64, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(softplus2): Softplus(beta=1, threshold=20)
)
)
(conv_to_fc): Linear(in_features=64, out_features=64, bias=True)
(conv_to_fc_softplus): Softplus(beta=1, threshold=20)
(fc_out): Linear(in_features=64, out_features=1, bias=True)
)なおモデルの詳細を綺麗な形で確認するなら「torchinfoモジュールのsummary」を使用します。
[IN]
from torchinfo import summary
# _df = graph_features(df_train.iloc[[1], :], atom_dict, 'train_feats(test)_1data.npz')
dataset = CIFDataset(df = _df, has_labels = True)
cf = CollateFn(has_labels = True)
batch = cf(dataset)
print(f'dataset数: {len(dataset)}, |batch Type{type(batch)} Keys数: {len(batch)}') #各KEYの中身を確認
print(f'atom_fea: type: {type(batch["atom_fea"])} shape: {batch["atom_fea"].shape}')
print(f'nbr_fea: type: {type(batch["nbr_fea"])} shape: {batch["nbr_fea"].shape}')
print(f'nbr_fea_idx: type: {type(batch["nbr_fea_idx"])} shape: {len(batch["nbr_fea_idx"])}')
print(f'crystal_atom_idx: type: {type(batch["crystal_atom_idx"])} shape: {len(batch["crystal_atom_idx"])}')
print(f'ids: type: {type(batch["ids"])} shape: {len(batch["ids"])}')
print(f'targets: type: {type(batch["targets"])} shape: {batch["targets"].shape}', end = '\n\n')
summary(model, input_data=(batch["atom_fea"].to(device, dtype = torch.long),
batch["nbr_fea"].to(device, dtype = torch.long),
batch["nbr_fea_idx"].to(device, dtype = torch.long),
batch["crystal_atom_idx"]))
[OUT]
dataset数: 1, |batch Type<class 'dict'> Keys数: 6
atom_fea: type: <class 'torch.Tensor'> shape: torch.Size([48])
nbr_fea: type: <class 'torch.Tensor'> shape: torch.Size([48, 12, 41])
nbr_fea_idx: type: <class 'torch.Tensor'> shape: 48
crystal_atom_idx: type: <class 'list'> shape: 1
ids: type: <class 'list'> shape: 1
targets: type: <class 'torch.Tensor'> shape: torch.Size([1])
==========================================================================================
Layer (type:depth-idx) Output Shape Param #
==========================================================================================
CrystalGraphConvNet -- --
├─ModuleList: 1-1 -- --
├─Embedding: 1-2 [48, 64] 5,696
├─ModuleList: 1-1 -- --
│ └─ConvLayer: 2-1 [48, 64] --
│ │ └─Linear: 3-1 [48, 12, 128] 21,760
│ │ └─BatchNorm1d: 3-2 [576, 128] 256
│ │ └─Sigmoid: 3-3 [48, 12, 64] --
│ │ └─Softplus: 3-4 [48, 12, 64] --
│ │ └─BatchNorm1d: 3-5 [48, 64] 128
│ │ └─Softplus: 3-6 [48, 64] --
│ └─ConvLayer: 2-2 [48, 64] --
│ │ └─Linear: 3-7 [48, 12, 128] 21,760
│ │ └─BatchNorm1d: 3-8 [576, 128] 256
│ │ └─Sigmoid: 3-9 [48, 12, 64] --
│ │ └─Softplus: 3-10 [48, 12, 64] --
│ │ └─BatchNorm1d: 3-11 [48, 64] 128
│ │ └─Softplus: 3-12 [48, 64] --
│ └─ConvLayer: 2-3 [48, 64] --
│ │ └─Linear: 3-13 [48, 12, 128] 21,760
│ │ └─BatchNorm1d: 3-14 [576, 128] 256
│ │ └─Sigmoid: 3-15 [48, 12, 64] --
│ │ └─Softplus: 3-16 [48, 12, 64] --
│ │ └─BatchNorm1d: 3-17 [48, 64] 128
│ │ └─Softplus: 3-18 [48, 64] --
├─Softplus: 1-3 [1, 64] --
├─Linear: 1-4 [1, 64] 4,160
├─Softplus: 1-5 [1, 64] --
├─Linear: 1-6 [1, 1] 65
==========================================================================================
Total params: 76,353
Trainable params: 76,353
Non-trainable params: 0
Total mult-adds (M): 3.87
==========================================================================================
Input size (MB): 0.19
Forward/backward pass size (MB): 3.64
Params size (MB): 0.31
Estimated Total Size (MB): 4.14
==========================================================================================5-10.学習・推論
モデルを学習させるために必要な関数・クラスを作成します。
評価指標を計算するために”RMSE”関数を作成
モデルを訓練させるために"training()"関数を作成
モデル検証時(学習やパラメータ更新は実施しない)用の関数を別途作成
交差検証用に"KFold()"インスタンスを作成して、DataFrameに検証時のラベリングを実施
値を標準化(平均0、分散:1)するためのクラスを作成
GPUを使用するために乱数固定、Device作成
[IN]
def RMSE(pred, target):
return np.sqrt(mean_squared_error(pred, target))
def training(df, model, criterion, device, normalizer, optimizer, epoch):
model.to(device) #モデルをGPUへ転送
model.train() #モデルを訓練モードに設定
#データセットの作成
train_dataset = CIFDataset(df = df, has_labels = True)
train_dataloader = DataLoader(train_dataset, batch_size = 256, shuffle = True, collate_fn = CollateFn(has_labels = True))
stream = tqdm(train_dataloader)
for batch_idx, batch in enumerate(stream, start = 1):
optimizer.zero_grad() #勾配の初期化
#順伝播(GPUへのデータの転送)
out_norm = model(
atom_fea = batch["atom_fea"].to(device, dtype = torch.long),
nbr_fea = batch["nbr_fea"].to(device, dtype = torch.long),
nbr_fea_idx = batch["nbr_fea_idx"].to(device, dtype = torch.long),
crystal_atom_idx = [atom_idx.to(device, dtype = torch.long) for atom_idx in batch["crystal_atom_idx"]]
)
target = batch["targets"]
target_norm = normalizer.norm(target).to(device, dtype = torch.float)
loss = criterion(out_norm, target_norm)#損失関数の計算
loss.backward() #逆伝播
optimizer.step() #パラメータの更新
out = normalizer.denorm(out_norm.detach().cpu()).numpy() #予測値の計算
stream.set_description("Epoch {:d} - train loss:{:.4f}".format(epoch, loss.item()))
return model
def infer(df, model, criterion, device, normalizer, has_labels = False, epoch = 1):
model.to(device) #モデルをGPUへ転送
model.eval() #モデルを推論モードに設定
#データセットの作成
infer_dataset = CIFDataset(df = df, has_labels = has_labels)
infer_dataloader = DataLoader(infer_dataset, batch_size = 256, shuffle = False, collate_fn = CollateFn(has_labels = has_labels))
stream = tqdm(infer_dataloader)
preds = []
for batch_idx, batch in enumerate(stream, start = 1):
with torch.no_grad():
pred_norm = model(
atom_fea = batch["atom_fea"].to(device, dtype = torch.long),
nbr_fea = batch["nbr_fea"].to(device, dtype = torch.long),
nbr_fea_idx = batch["nbr_fea_idx"].to(device, dtype = torch.long),
crystal_atom_idx = [atom_idx.to(device, dtype = torch.long) for atom_idx in batch["crystal_atom_idx"]]
)
pred = normalizer.denorm(pred_norm).detach().cpu().numpy().ravel()
if has_labels:
target = batch["targets"]
target_norm = normalizer.norm(target).to(device, dtype = torch.float)
loss = criterion(pred_norm, target_norm)
stream.set_description("Epoch {:d} - validate loss:{:.4f}".format(epoch, loss.item()))
preds.append(pred)
preds = np.concatenate(preds, axis = 0)
return preds
kf = KFold(n_splits = 5, shuffle = True, random_state = 42)
for i, (train_index, valid_index) in enumerate(kf.split(df_train)):
df_train.loc[valid_index, "fold"] = i
df_train.head()
[OUT]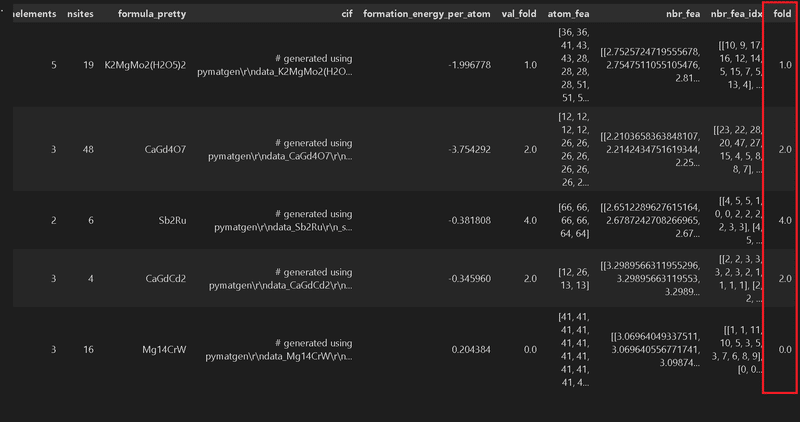
[IN]
class Normalizer(object):
"""Normalize a Tensor and restore it later. """
def __init__(self, tensor):
"""tensor is taken as a sample to calculate the mean and std"""
self.mean = torch.mean(tensor)
self.std = torch.std(tensor)
def norm(self, tensor):
return (tensor - self.mean) / self.std
def denorm(self, normed_tensor):
return normed_tensor * self.std + self.mean
def state_dict(self):
return {'mean': self.mean,
'std': self.std}
def load_state_dict(self, state_dict):
self.mean = state_dict['mean']
self.std = state_dict['std']
[OUT][IN]
def seed_everything(seed=42):
np.random.seed(seed%(2**32-1))
random.seed(seed)
torch.manual_seed(seed)
torch.cuda.manual_seed(seed)
torch.backends.cudnn.deterministic =True
torch.backends.cudnn.benchmark = False
seed_everything()
# device optimization
if torch.cuda.is_available():
device = torch.device("cuda")
else:
device = torch.device("cpu")
print(f"Using device: {device}")
[OUT]
Using device: cuda準備が出来たらいつもの流れで学習させます。
(線形)モデルを使用して推論値を計算する
損失関数を使用して"正解値"と"推論値"から誤差を求める
loss.backward()で各パラメータの勾配を求める
optimizer.step()(または手動)によりパラメータを更新する
zero_grad()によりパラメータの勾配を初期化する
1~5をループ計算する
[IN]
optimizer = torch.optim.Adam(params = model.parameters(), lr = 1e-2)
scheduler = StepLR(optimizer, step_size = 1, gamma = 0.1, verbose = True)
criterion = nn.MSELoss()
fold = 0
best_rmse = np.inf
train_targets = torch.Tensor(df_train.loc[df_train["fold"] != fold, 'formation_energy_per_atom'].to_numpy())
normalizer = Normalizer(train_targets)
for epoch in range(1, 5):
model = training(df_train.loc[df_train["fold"] != fold], model, criterion, device, normalizer, optimizer, epoch)
preds = infer(df_train.loc[df_train["fold"] == fold], model, criterion, device, normalizer, has_labels = True, epoch = epoch)
epoch_rmse = RMSE(preds, df_train.loc[df_train["fold"] == fold, 'formation_energy_per_atom'])
scheduler.step()
print("Epoch {:d} - validate RMSE: {:.4f}".format(epoch, epoch_rmse))
if best_rmse > epoch_rmse:
model_path = "{:s}/cgcnn_fold{:d}.pth".format('model', fold)
torch.save(model.state_dict(), model_path)
print("model {:s} saved.".format(model_path))
best_rmse = epoch_rmse
[OUT]
Epoch 1 - validate RMSE: 1.1969
Epoch 2 - validate RMSE: 1.1969
Epoch 3 - validate RMSE: 1.1970
Epoch 4 - validate RMSE: 1.19715ー11.提出用(テスト)データの推論
最後にテストデータを推論します。
[IN]
filename = "test_feats.npz"
feats_filepath = os.path.join('feats', filename)
keys = ["atom_fea", "nbr_fea", "nbr_fea_idx"]
if os.path.exists(feats_filepath):
graph_feats = np.load(feats_filepath, allow_pickle=True)
for k in keys:
df_test[k] = graph_feats[k]
else:
test_df = graph_features(df_test, atom_dict, filename, max_num_nbr = 12, radius = 8)
#提出用のデータセット読み込み
sub_df = df_submit.copy()
#modelsフォルダに保存したモデルを読み込み
preds = infer(df_test, model, criterion, device, normalizer, has_labels = False)
sub_df["formation_energy_per_atom"] = preds
sub_df.to_csv("output/submission_CGCNN(test).csv", index = False)
[OUT]5-12.提出・結果確認
最後に指定の方式に変更してサイトから提出します。結果はLightGBMの方が優れていました。Nishikaより下記見解で性能が低いと予想されます。
==================================
今回コンペデータを試行した学習は収束しておりません。CGCNNの論文では使用するデータについて収束しない個体を除いておりますが、今回は検討しておらず学習が収束していない原因である可能性があります。
==================================

参考資料
あとがき
その2を出す時間を作らないと・・・とりあえず次はAutoMLと全結合で処理する。
この記事が気に入ったらサポートをしてみませんか?