SARS-Cov-2に対する抗体についての論文(名大らの研究)

9/19にtwitterに↓の記事があったので、記事のもととなった論文を見てみました。

発表された論文はこちら→https://advances.sciencemag.org/content/early/2020/09/18/sciadv.abd3916/tab-pdf
いつも通り、google翻訳に頼り切ってます。注釈などは省略しました。
・・・・・・・・・・・・・・
Antibody-like proteins that capture and neutralize SARSCoV-2
SARSCoV-2を捕捉して中和する抗体様タンパク質
To combat SARS-CoV-2 and any unknown emerging pathogens in the future, the development of a rapid and effective method to generate high-affinity antibodies or antibody-like proteins is of critical importance.
SARS-CoV-2および将来の未知の病原菌と戦うためには、高親和性抗体または抗体様タンパク質を生成するための迅速で効果的な方法の開発が非常に重要です。
We here report a high-speed in vitro selection of multiple high-affinity antibody-like proteins against various targets including the SARS-CoV-2 spike protein.
ここでは、SARS-CoV-2スパイクタンパク質を含むさまざまなターゲットに対する複数の高親和性抗体様タンパク質の高速in vitro選択について報告します。
※in vitro=体外・試験管内と訳されている。整えられた実験環境のこと。
The sequences of monobodies against the SARSCoV-2 spike protein were successfully procured within only four days.
SARSCoV-2スパイクタンパク質に対するモノボディのシーケンスは、わずか4日以内に正常に取得されました。
Furthermore, the obtained monobody efficiently captured SARS-CoV-2 particles from the nasal swab samples of patients and exhibited a high neutralizing activity against SARS-CoV-2 infection (IC50 = 0.5 nM).
さらに、得られたモノボディは、患者の鼻腔スワブサンプルから効果的にSARS-CoV-2粒子を捕捉し、SARS-CoV-2感染に対して高い中和活性を示しました(IC50 = 0.5 nM)。
The high-speed in vitro selection of antibody-like proteins would be useful for the rapid development of a detection method and a neutralizing protein against a virus responsible for an ongoing, and possibly a future, pandemic.
抗体様タンパク質の高速in vitro選択は、検出方法の迅速な開発と、進行中の、場合によっては将来のパンデミックの原因となるウイルスに対する中和タンパク質の開発に役立ちます。
Introduction
The coronavirus disease 2019 (COVID-19) pandemic, which was caused by the severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2), has negatively and deeply affected our lives and societies.
重症急性呼吸器症候群関連コロナウイルス2(SARS-CoV-2)が原因で発生したコロナウイルス疾患2019(COVID-19)のパンデミックは、私たちの生活や社会に悪影響を及ぼし、深刻な影響を及ぼしています。
To control the COVID-19 pandemic in addition to any emerging, heretofore unknown pathogens in the future, it is of paramount importance to rapidly generate multiple high-affinity antibodies or antibody-like proteins against virus proteins, thus developing both a detection method for the virus(1) and a neutralizing protein against it(2).
COVID-19および将来発生する未知の病原菌のパンデミックを制御するには、ウイルスタンパク質に対して複数の高親和性抗体または抗体様タンパク質を迅速に生成することが最も重要です。したがって、ウイルスの検出方法とそれに対する中和タンパク質の両方を開発しています。
The identification of a cross-reactive antibody among those developed against SARS-CoV is the fastest way to obtain an antibody(3-7).
SARS-CoVに対して開発された抗体の中から交差反応性抗体を特定することは、抗体を入手する最も早い方法です。
In principle, however, this approach would yield lower-affinity antibodies with cross-reactivity to the original virus.
ただし、原則として、このアプローチでは、元のウイルスに対する交差反応性を持つ親和性の低い抗体が生成されます。
The isolation of monoclonal antibodies from virus-infected patients is another possible approach, but the collection of B cell samples and the identification and large-scale production of effective antibodies represent a long-term research and development project.
ウイルスに感染した患者からモノクローナル抗体を分離することも考えられるアプローチの1つですが、B細胞サンプルの収集と効果的な抗体の同定と大量生産は、長期的な研究開発プロジェクトです。
In vitro selection is advantageous in terms of isolation speed of antibody-like proteins (ALPs) because time-consuming animal immunization is not necessary to generate ALPs.
ALPを生成するために時間のかかる動物の免疫化が必要ないため、in vitro選択は抗体様タンパク質(ALP)の分離速度の点で有利です。
This method also allows the use of immunoglobulin protein scaffolds, including single-chain variable fragments, single-domain antibodies, and non-immunoglobulin proteins.
この方法では、単鎖可変フラグメント、単一ドメイン抗体、非免疫グロブリンタンパク質などの免疫グロブリンタンパク質足場を使用することもできます。
Such non-immunoglobulin proteins as a lipocalin, a domain of fibronectin, the Z-domain of protein A, the ankyrin repeat motif, and SH3 domain of Fyn allow a greater chance to obtain high-affinity ALPs.
リポカリン、フィブロネクチンのドメイン、プロテインAのZドメイン、アンキリンリピートモチーフ、FynのSH3ドメインなどの非免疫グロブリンタンパク質は、高親和性ALPを取得する可能性が高くなります。
Phage display is the method that is used most commonly for in vitro selection.
ファージディスプレイは、in vitroでの選択に最も一般的に使用される方法です。
※ファージディスプレイ(Phage display):タンパク質間相互作用あるいはタンパク質とその他標的物質との相互作用を検出する方法の一つ。重要な応用として、多数の遺伝子を含むライブラリの中から特定の標的に結合するものを選抜・濃縮することができる。これはタンパク質の人工的”進化”にも応用でき、自然選択になぞられてインビトロセレクション(In vitro selection)と呼ばれている。
ALP libraries were expressed in E.coli and displayed on the surface protein of phage.
ALPライブラリーは大腸菌で発現され、ファージの表面タンパク質に表示されました。
The E. coli transformation step using phage DNA, the efficiency of which limited the library size practically to 109 to 1011, is the main disadvantage of phage display.
ファージDNAを使用した大腸菌の形質転換ステップでは、ライブラリーのサイズが実質的に109〜1011に制限されていましたが、これはファージディスプレイの主な欠点です。
To generate highly diverse protein libraries, typically 1012 to 1013, a cell-free translation system has been used for mRNA display.
非常に多様なタンパク質ライブラリ(通常は1012〜1013)を生成するために、無細胞翻訳システムがmRNAディスプレイに使用されています。
However, this display method requires multistep manipulations to synthesize protein/mRNA/puromycin-linker (PuL) complexes;the transcription of the mRNA; the attachment of the PuL to the mRNA; and the translation of library proteins.
ただし、この表示方法では、タンパク質/ mRNA /ピューロマイシン-リンカー(PuL)複合体;mRNAの転写; mRNAへのPuLの付着; ライブラリータンパク質の翻訳を合成するために多段階の操作が必要です。
These multistep manipulations render mRNA display a time-consuming method, with more than weeks typically being necessary to complete six to seven rounds of the procedure (2–3 days per round).
これらの多段階の操作により、mRNAは時間のかかる方法で表示されます。通常、手順の6〜7ラウンド(ラウンドあたり2〜3日)を完了するには数週間以上必要です。
We previously modified the mRNA display method and developed a high-speed in vitro selection method, i.e., the transcription–translation coupled with association of puromycin linker (TRAP) display, for the identification of macrocyclic peptides (Fig. S1A).
以前に、mRNA表示方法を変更し、大環状ペプチドを同定するための高速in vitro選択方法、つまり、転写-翻訳とピューロマイシンリンカー(TRAP)表示の関連付けを開発しました(図S1A)。
In the TRAP display, macrocyclic peptide/mRNA/PuL complexes are automatically synthesized via the simple addition of the peptide template DNA, thus skipping two time-consuming steps: the transcription of mRNA and the ligation of PuL.
TRAP表示では、大環状ペプチド/ mRNA / PuL複合体は、ペプチドテンプレートDNAを追加するだけで自動的に合成されます。したがって、時間のかかる2つのステップ(mRNAの転写とPuLのライゲーション)をスキップします。
We reported the completion of six rounds of selection in approximately 14 hours using TRAP display, and obtained macrocyclic peptides with nanomolar affinity to their target protein.
TRAPディスプレイを使用して約14時間で6ラウンドの選択が完了したことを報告し、標的タンパク質に対してナノモルの親和性を持つ大環状ペプチドを得た。
We also performed selection against the vascular endothelial growth factor receptor 2 (VEGFR2), and obtained a macrocyclic peptide with VEGF-induced angiogenesis inhibitory activity.
また、血管内皮増殖因子受容体2(VEGFR2)に対する選択を行い、VEGFが誘導する血管新生阻害活性を持つ大環状ペプチドを得ました。
Here, we report the development of an improved TRAP display to facilitate the selection of ALPs (Fig. 1A and Fig.S1A).
ここでは、ALPの選択を容易にするために改善されたTRAPディスプレイの開発について報告します(図1Aおよび図S1A)。
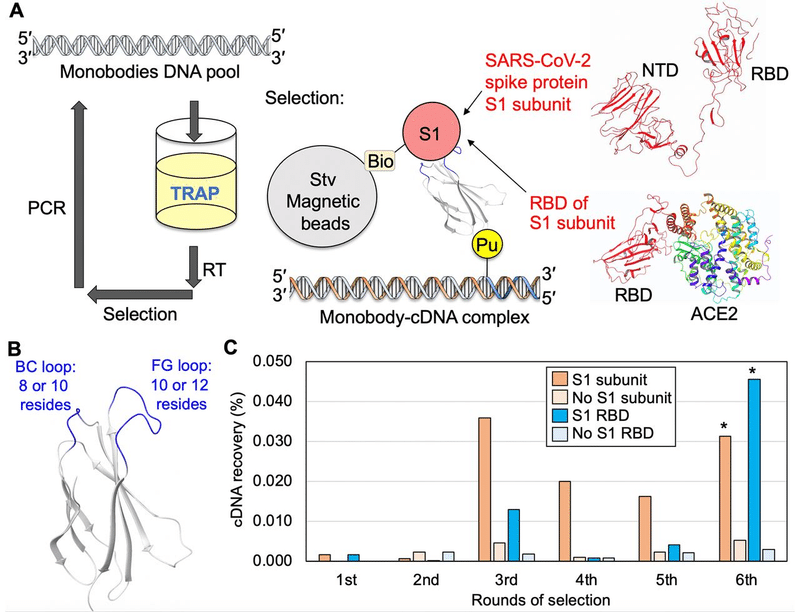
Subsequently, we performed test selections against the human epidermal growth factor receptor 1 (EGFR1) and the epithelial growth factor receptor type 2 (HER2) using a nanobody (the camelid single-domain antibody) and a monobody (the 10th type III domain of human fibronectin) as backbone proteins.
続いて、人間の上皮成長因子受容体1(EGFR1)と上皮成長因子受容体タイプ2(HER2)に対して、ナノボディ(ラクダ科の単一ドメイン抗体)とモノボディ(ヒトフィブロネクチンの10番目のタイプIIIドメイン)をバックボーンタンパク質として使用してテスト選択を行いました。
We further used this method to rapidly obtain monobodies against the S1 subunit of SARSCoV-2 spike protein.
さらに、この方法を使用して、SARSCoV-2スパイクタンパク質のS1サブユニットに対するモノボディを迅速に取得しました。
The selected high-affinity monobodies were used to capture SARS-CoV-2 particles from nasal swab samples of patients.
選択した高親和性モノボディを使用して、患者の鼻腔スワブサンプルからSARS-CoV-2粒子をキャプチャしました。
We also increased the affinity of a monobody by the dimerization.
また、二量体化によりモノボディの親和性を高めました。
The monomeric and dimeric monobodies were used as a neutralizing protein against SARS-CoV2 infection.
単量体および二量体モノボディは、SARS-CoV2感染に対する中和タンパク質として使用されました。
Results
Development of the improved TRAP display for antibody-like protein selection We started our study by measuring the display efficiencies of the nanobody and monobody on the mRNA, because they determine the diversity of ALP pools and the selection efficiency against a target protein.
抗体様タンパク質選択のための改善されたTRAP表示の開発調査は、標的タンパク質に対するALPプールの多様性と選択効率を決定するため、mRNA上のナノボディとモノボディの表示効率を測定することから始めました。
The monobody DNA template and the DNA-puromycin linker (dPuL, Fig. S1B) were added to the TRAP system, followed by the determination of the display efficiency of the monobody on the mRNA.
モノボディDNAテンプレートとDNAピューロマイシンリンカー(dPuL、図S1B)をTRAPシステムに追加し、mRNAでのモノボディの表示効率を測定しました。
In contrast with our expectations, less than 3% of the dPuL formed a monobody/mRNA/dPuL complex;
予想とは対照的に、dPuLの3%未満がモノボディ/ mRNA / dPuL複合体を形成しました。
moreover, most of the dPuL (87%) did not form an mRNA/dPuL complex (Fig. S1C).
さらに、ほとんどのdPuL(87%)はmRNA / dPuL複合体を形成しませんでした(図S1C)。
As an mRNA sufficient to capture the dPuL (1 μM) was produced within 10 min in the reaction mixture (Fig. S2A), an unexpected reaction occurred that prohibited the formation of the mRNA/dPuL complex.
dPuL(1μM)を捕捉するのに十分なmRNAが反応混合物中で10分以内に生成されたため(図S2A)、mRNA / dPuL複合体の形成を妨げる予期しない反応が発生しました。
We found that the DNA part of the dPuL that was complementary to the 21mers of the 3′-end mRNA sequence (Fig. S1B) was transcribed to RNA in a promoter-independent manner, and formed an undesired DNA/RNA duplex;
3 '末端のmRNAシーケンスの21 merに相補的なdPuLのDNA部分(図S1B)が、プロモーター非依存的な方法でRNAに転写され、望ましくないDNA / RNA二重鎖を形成することがわかりました
therefore, it inhibited the formation of the mRNA/dPuL complex (Fig. S2B).
したがって、mRNA / dPuL複合体の形成を阻害しました(図S2B)。
To prevent promoter-independent transcription, we added a 2′-methoxy (OMe) modification on the ribose because it was reported to prevent T7 promoter-dependent transcription.
プロモーター非依存の転写を防ぐために、リボースに2'-メトキシ(OMe)修飾を追加しました。これは、T7プロモーター依存の転写を防ぐことが報告されているためです。
Indeed, the 2′-OMe modification also prevented the promoter-independent transcription, even after an incubation of 30 min (Fig. S2B).
実際、2'-OMeの修飾により、30分のインキュベーション後でも、プロモーターに依存しない転写が妨げられました(図S2B)。
We then used the 2′-methoxy-modified PuL (mPuL) in the TRAP display.
次に、TRAPディスプレイで2'-メトキシ修飾PuL(mPuL)を使用しました。
As expected, the efficiency of the formation of the mRNA/mPuL complex was increased, from 11% (dPuL) to 60% (mPuL), and the display efficiency was augmented, from 3% (dPuL) to 22% (mPuL) (Fig. S1C).
予想どおり、mRNA / mPuL複合体の形成効率が11%(dPuL)から60%(mPuL)に増加し、表示効率が3%(dPuL)から22%(mPuL)に増加しました(図S1C)。
We also analyzed the display efficiency of the anti-triclocarban nanobody on the mRNA.
また、mRNAに対する抗トリクロカルバンナノボディの表示効率も分析しました。
The efficiency of the formation of the mRNA/mPuL complex was increased, from 9% (dPuL) to 45% (mPuL);
mRNA / mPuL複合体の形成効率が9%(dPuL)から45%(mPuL)に向上しました。
moreover, the display efficiency of the nanobody on the mRNA was also increased, from 2% (dPuL) to 15% (mPuL) (Fig. S1C).
さらに、mRNAに対するナノボディの表示効率も2%(dPuL)から15%(mPuL)に増加しました(図S1C)。
The TRAP display was very useful to simplify the selection procedure, but it was not ideal for the first-round selection, because the diversity of the library was limited by the amount of the DNA template added in the translation mixture (see Fig. S2C for the optimization of DNA concentration).
TRAP表示は、選択手順を簡略化するのに非常に役立ちましたが、ライブラリの多様性は、翻訳混合物に追加されたDNAテンプレートの量によって制限されたため、初回の選択には理想的ではありませんでした(DNA濃度の最適化については、図S2Cを参照してください)。
To maximize the diversity of libraries, we usually stock an mRNA pool and add it to the translation reaction after the formation of the mRNA/PuL complex in the first-round selection.
ライブラリーの多様性を最大化するために、通常、mRNAプールをストックし、最初のラウンドの選択でmRNA / PuL複合体が形成された後、それを翻訳反応に追加します。
Therefore, we also analyzed the display efficiency of the anobody/monobody on the mRNA in the first-round format.
したがって、最初のラウンドの形式で、mRNAに対するアノボディ/モノボディの表示効率も分析しました。
We found that the display efficiencies were similar to that of the TRAP display format for both the monobody (19%) and nanobody (15%) (Fig. S1D).
表示効率は、モノボディ(19%)とナノボディ(15%)の両方で、TRAP表示フォーマットと同様であることがわかりました(図S1D)。
Next, we conducted a test selection using the monobody and nanobody libraries against EGFR1 and HER2 (Fig. S3).
次に、EGFR1とHER2に対するモノボディとナノボディのライブラリを使用してテスト選択を行いました(図S3)。
High-affinity binders (KD = 1.3 nM and 3.5 nM for EGFR1, and KD = 4.4 nM and 13 nM for HER2) were obtained from the monobody library, whereas mid-affinity ones were obtained from the nanobody library (KD = 47 nM and 32 nM for EGFR1, and KD = 11 nM and 9.9 nM for HER2).
High-affinity binders were obtained from the monobody library, whereas mid-affinity ones were obtained from the nanobody library (KD = 47 nM and 32 nM for EGFR1, and KD = 11 nM and 9.9 nM for HER2).
高親和性バインダーはモノボディライブラリ(EGFR1:KD = 1.3 nMおよび3.5 nM、HER2:KD = 4.4 nMおよび13 nM)から取得されましたが、中親和性バインダーはナノボディライブラリ(EGFR1:KD = 47 nMおよび32 nM、HER2:KD = 11 nMおよび9.9 nM)から取得されました。
High-speed selection of monobodies against the SARSCoV-2 spike protein S1 subunit
SARSCoV-2スパイクタンパク質S1サブユニットに対するモノボディの高速選択
According to the results presented above, we focused on the monobody for the subsequent library construction.
上記の結果から、その後のライブラリ構築では、モノボディに焦点を当てました。
We introduced 8 or 10 random residues at the BC loop (Pro25–Val29) and 10 or 12 random residues at the FG loop (Gly77–Lys86) of the monobody (Fig. 1B), according to previous reports.
以前のレポートによると、モノボディのBCループ(Pro25–Val29)に8または10個のランダム残基を導入し、FGループ(Gly77–Lys86)に10個または12個のランダム残基を導入しました(図1B)。
We used a Tyr-, Ser-, Gly-, and Trp-rich codon mix to construct the random residues.
Tyr、Ser、Gly、およびTrpが豊富なコドンミックスを使用して、ランダムな残基を構築しました。
※Tyr=トレオニン Ser=セリン Gly=グリシン Trp=トリプトファン
The preparation of the mRNA library took 6 months after we ordered the oligonucleotides, because of the oligonucleotide synthesis using trinucleotide phosphoramidites(29) and the large-scale PCR and transcription procedures that were applied to maintain the diversity of the monobody library.
オリゴヌクレオチドの注文後、mRNAライブラリの準備に6か月かかりました。これは、トリヌクレオチドホスホラミダイトを使用したオリゴヌクレオチド合成と、モノボディライブラリの多様性を維持するために適用された大規模なPCRおよび転写手順のためです。
However, once the pool of mRNA was prepared, it could be used for multiple in vitro selection studies against various targets because of the large amount of mRNA produced.
ただし、mRNAのプールが準備されると、生成されるmRNAの量が多いため、さまざまなターゲットに対する複数のin vitro選択研究に使用できます。
With the pool mRNA in our hands, we received the two biotinylated targets, the SARS-CoV-2 spike protein S1 subunit (S1-biotin) and the receptor binding domain (RBD-biotin), and started the TRAP display selection (Fig. 1A).
私たちはプールmRNAを手に入れ、ビオチン化された2つのターゲット、SARS-CoV-2スパイクタンパク質S1サブユニット(S1-ビオチン)と受容体結合ドメイン(RBD-ビオチン)を受け取り、TRAP表示の選択を開始しました(図1A)。
In the first round of the selection, an mRNA template, rather than a DNA one, was used to maximize the diversity of the monobody library (1 μM mRNA in a 500 μL scale; monobody display efficiency on the mRNA of about 10%; 3 × 1013 calculated molecules; Fig. S4).
選択の最初のラウンドでは、モノボディライブラリーの多様性を最大化するために、DNAではなくmRNAテンプレートが使用されました(500μLスケールで1μMmRNA;約10%のmRNAでのモノボディ表示効率; 3×1013計算分子;図S4)。
We used mixed targets in the first round, to optimize the effort of the large-scale preparation of the cell-free translation system.
最初のラウンドでは混合ターゲットを使用して、無細胞翻訳システムの大規模な準備の労力を最適化しました。
From the second round, we used the TRAP display to boost the speed of selection, and simultaneously conducted selection procedures against each target.
第2ラウンドからは、TRAPディスプレイを使用して選択の速度を高め、同時に各ターゲットに対して選択手順を実行しました。
We observed an enrichment of binder monobodies against both targets in the third round of selection (Fig. 1C).
3回目の選択では、両方のターゲットに対してバインダーモノボディの濃縮が観察されました(図1C)。
To obtain higher-affinity monobodies, we performed an additional three rounds of selection using a 10-times lower target concentration (2 nM).
より高い親和性のモノボディを得るために、10倍低いターゲット濃度(2 nM)を使用して、さらに3回の選択を行いました。
After the six rounds of selection, the recovered cDNAs of monobodies were sequenced.
6ラウンドの選択の後、回収されたモノボディのcDNAが配列決定されました。
Notably, because of the high-speed in vitro selection feature of the TRAP display (Fig. 1A), we successfully completed the selection within 3 days, and obtained the monobody sequences on the fourth day.
特に、TRAPディスプレイの高速in vitro選択機能(図1A)のため、3日以内に選択を正常に完了し、4日目にモノボディシーケンスを取得しました。
Receptor binding domain-specific monobodies with low- to sub-nM KD values
低〜サブnMのKD値を持つ受容体結合ドメイン固有のモノボディ
As the same clones were enriched through the selection against both the S1 subunit and the RBD, we selected the nine most-enriched clones in the pool (Table 1) for further study.
同じクローンがS1サブユニットとRBDの両方に対する選択によって濃縮されたので、さらに調査するために、プール内で最も濃縮された9つのクローンを選択しました(表1)。
To confirm the binding activity, we immobilized the purified monobodies (Table S1, Fig. S5) on a microwell plate and added either the S1 subunit (S1–HRP) or the RBD conjugated with streptavidin-horseradish peroxidase (Stv-HRP).
結合活性を確認するために、精製したモノボディ(表S1、図S5)をマイクロウェルプレートに固定化し、S1サブユニット(S1-HRP)またはストレプトアビジン-ホースラディッシュペルオキシダーゼ(Stv-HRP)とコンジュゲートしたRBDを追加しました。
The results of the enzyme-linked immunosorbent assay (ELISA) showed that the all monobodies were able to bind to both targets (Fig. 2A).
酵素結合免疫吸着測定法(ELISA)の結果は、すべてのモノボディが両方のターゲットに結合できることを示しました(図2A)。
In fact, the wild-type monobody (WT, the control protein with the original loop sequences) did not bind to either of the targets.
実際、野生型モノボディ(WT、元のループシーケンスを持つコントロールタンパク質)はどちらのターゲットにも結合しませんでした。
These results clearly indicated that the selected loop sequences were responsible for the binding to the RBD of the S1 subunit.
これらの結果は、選択されたループシーケンスがS1サブユニットのRBDへの結合に関与していることを明確に示しています。
To study the stability of monobodies, C-terminally biotinylated monobodies prepared by sortase A reaction (monobody-biotin, Fig. S5) were incubated overnight at various temperatures, and the remaining binding activity was measured by ELISA using an S1 subunit-immobilized microplate and Stv-HRP.
モノボディの安定性を研究するために、ソルターゼA反応によって調製されたC末端ビオチン化モノボディ(モノボディビオチン、図S5)をさまざまな温度で一晩培養しました。そして、残りの結合活性は、S1サブユニット固定化マイクロプレートとStv-HRPを使用してELISAで測定しました。
We found that all monobodies were stable at 37°C, and that five of them retained half of the binding activity after incubation at 50°C overnight (Fig. S6).
すべてのモノボディは37°Cで安定しており、そのうちの5つは50°Cで一晩培養した後も結合活性の半分を保持していることがわかりました(図S6)。
We further assessed the kinetic parameters of all nine monobodies using bio-layer interferometry (BLI).
さらに、生体層干渉法(BLI)を使用して、9つのモノボディすべての動態パラメータを評価しました。
S1-biotin was immobilized on the streptavidin-immobilized sensor and various concentrations of monobodies were added, to determine the kinetic parameters based on global fitting (Fig. 2B,Table 1).
S1-ビオチンがストレプトアビジン固定化センサーに固定化され、さまざまな濃度のモノボディが追加されて、グローバルフィッティングに基づいて速度論的パラメーターが決定されました(図2B、表1)。
The seven clones exhibited a sub-nM to nM-level affinity against the S1 subunit (clone 1, KD = 2.47 nM; clone 4, KD = 1.94 nM; clone 6, KD = 0.76 nM; clone 10, KD = 2.02 nM; clone 11, KD = 3.23 nM; clone 12, KD = 1.37 nM; clone 18, KD = 0.65 nM).
7つのクローンは、S1サブユニットに対してサブnMからnMレベルの親和性を示しました(クローン1、KD = 2.47 nM、クローン4、KD = 1.94 nM、クローン6、KD = 0.76 nM、クローン10、KD = 2.02 nM、クローン11、KD = 3.23 nM、クローン12、KD = 1.37 nM、クローン 18、KD = 0.65 nM)。
Since mutations were found in the clone 6 (S55W), clone 11 (L62M and S to T in the BC loop), and clone 12 (D67H), we also determined the kinetic parameters of the back mutants (6b, 11b, and 12b).
変異はクローン6 (S55W)、クローン11 (L62M and S to T in the BC loop)、およびクローン12 (D67H)で見つかったため、バック変異体の速度論的パラメーターも決定しました(6b, 11b, and 12b)。
The affinities were similar to those observed for the parental ones, suggesting that these mutations do not contribute to the binding affinity.
親和性は親の親和性で観察されたものと同様であり、これらの変異は結合親和性に寄与しないことを示唆しています。
It was interesting to obtain high-affinity monobodies, because the in vitro selection of ALPs sometimes required additional affinity maturation after the initial selection to obtain high-affinity monobodies.
ALPのin vitro選択では、高親和性モノボディを取得するために、最初の選択後に追加の親和性成熟が必要な場合があるため、高親和性モノボディを取得することは興味深いことでした。
We believe that this finding is attributable to either the high antigenicity of the S1 subunit or the high diversity/quality of the library prepared using trinucleotide phosphoramidite synthesis.
この発見は、S1サブユニットの高い抗原性、またはトリヌクレオチドホスホラミダイト合成を使用して調製されたライブラリーの高い多様性/品質に起因すると考えられます。
Monobodies bound to the SARS-CoV-2 spike protein S1 subunit but not to that of SARS-CoV
モノボディはSARS-CoV-2スパイクタンパク質S1サブユニットに結合しましたが、SARS-CoVのサブユニットには結合しませんでした
Next, we studied the specificity of the monobodies for the SARS-CoV-2 spike protein S1 subunit, because this parameter is important for the development of a future diagnostic assay for this virus.
次に、SARS-CoV-2スパイクタンパク質S1サブユニットのモノボディの特異性を調査しました。これは、このパラメーターがこのウイルスの将来の診断アッセイの開発に重要であるためです。
Since the SARS-CoV-2 spike protein sequence exhibits greater similarity with that of SARS CoV than it does with other human coronavirus, we decided to test the specificity of the monobodies against the S1 subunit of SARSCoV-2 and SARS-CoV.
SARS-CoV-2スパイクタンパク質配列は、他のヒトコロナウイルスよりもSARS CoVの類似性が高いため、SARSCoV-2およびSARS-CoVのS1サブユニットに対するモノボディの特異性をテストすることにしました。
The pull-down experiment using monobody-immobilized beads showed that four monobodies (clones 4, 6, 9, and 10) bound to the SARS-CoV-2 S1 subunit, but not to the SARS-CoV S1 subunit (Fig. 3A), suggesting that these monobodies are specific for the SARS-CoV-2 S1 subunit.
The pull-down experiment using monobody-immobilized beads showed that four monobodies (clones 4, 6, 9, and 10) bound to the SARS-CoV-2 S1 subunit, but not to the SARS-CoV S1 subunit, suggesting that these monobodies are specific for the SARS-CoV-2 S1 subunit.
モノボディ固定化ビーズを使用したプルダウン実験では、4つのモノボディ(クローン4、6、9、10)がSARS-CoV-2 S1サブユニットに結合しているが、SARS-CoV S1サブユニットには結合していないことが示された(図3A)。これらのモノボディはSARS-CoV-2 S1サブユニットに固有であることを示唆しています。
In turn, four monobodies (clones 1, 11, 12, and 18) bound to both targets, and clone 16 did not bind to either of the S1 subunits because of a low binding affinity (Table 1).
次に、4つのモノボディ(クローン1、11、12、18)が両方のターゲットに結合し、クローン16は結合親和性が低いため、どちらのS1サブユニットにも結合しませんでした(表1)。
Clone 16 pulled down a spike protein trimer (Fig. 3A), probably because of a multivalent effect of the target.
クローン16はスパイクプロテイントリマーを引き下げました(図3A)。これは、おそらくターゲットの多価効果によるものです。
Monobodies inhibited the interaction between ACE2 and the S1 subunit
モノボディはACE2とS1サブユニット間の相互作用を阻害した
To determine whether the binding site of the monobodies overlapped with that of the angiotensin-converting enzyme 2 (ACE2), we also tested the inhibition of the S1 subunit/ACE2 interaction afforded by the monobodies.
モノボディの結合部位がアンジオテンシン変換酵素2(ACE2)の結合部位と重複しているかどうかを判断するために、モノボディによって提供されるS1サブユニット/ ACE2相互作用の阻害もテストしました。
The S1 subunit was immobilized on a microplate and the pre-mixed solution of ACE2–HRP and each monobody was added.
S1サブユニットがマイクロプレートに固定化され、ACE2–HRPと各モノボディの事前混合溶液が追加されました。
The results of ELISA showed that monobody clones 4, 6, 9, and 10 at a concentration of 100 nM inhibited the binding of the S1 subunit to ACE2 (Fig. 3B);
ELISAの結果は、100 nMの濃度のモノボディクローン4、6、9、および10がS1サブユニットのACE2への結合を阻害することを示しました(図3B)。
moreover, the inhibition was retained for three of the monobodies (clones 4, 6, and 10) at the low concentration of 10 nM (Fig. 3C).
さらに、阻害は10 nMの低濃度で3つのモノボディ(クローン4、6、および10)に対して保持されました(図3C)。
Interestingly, these monobodies also exhibited specificity toward the SARS-CoV-2 S1 subunit (Fig. 3A), probably because the ACE2 binding subdomain of the RBD contains more substitutions than other regions of the spike protein.
興味深いことに、これらのモノボディはSARS-CoV-2 S1サブユニットに対しても特異性を示しました(図3A)。これは、RBDのACE2結合サブドメインに、スパイクタンパク質の他の領域よりも多くの置換が含まれているためと考えられます。
Application of monobodies in sandwich ELISA
サンドイッチELISAにおけるモノボディの適用
The nine monobodies studied above exhibited binding activity toward the S1 subunit.
上記の9つのモノボディは、S1サブユニットに対して結合活性を示しました。
This observation led us to develop a sandwich ELISA for the detection of the S1 subunit, because it requires a pair of antibodies that recognize distinct epitopes of the S1 subunit.
この観察により、S1サブユニットの検出用のサンドイッチELISAを開発することになりました。S1サブユニットの異なるエピトープを認識する抗体のペアが必要だからです。
To explore for such monobody pairs, we tested all combinations of the cloned monobodies in a sandwich ELISA format and found that the monobodies recognized three different epitopes (Fig. 3D, Table 1).
そのようなモノボディのペアを探索するために、サンドイッチELISA形式でクローン化モノボディのすべての組み合わせをテストし、モノボディが3つの異なるエピトープを認識することがわかりました(図3D、表1)。
Monobody clones 1, 11, 12, and 18 shared an epitope that was not located at the ACE2-binding surface (Figs. 3B and 3D).
モノボディクローン1、11、12、および18は、ACE2結合表面に配置されなかったエピトープを共有していました(図3Bおよび3D)。
The FG loop sequences of these monobodies were similar (xLWGYxTxWD; with x representing a non-conserved residue), suggesting that they are the main antigen-binding loop.
これらのモノボディのFGループシーケンスは類似しており(xLWGYxTxWD、xは保存されていない残基を表す)、これらがメインの抗原結合ループであることを示唆しています。
Monobody clones 6 and 10 shared another epitope that was likely located in the ACE2-binding subdomain (Figs. 3B and 3D).
モノボディクローン6と10は、ACE2結合サブドメインにある可能性が高い別のエピトープを共有していました(図3Bと3D)。
These clones exhibited a conserved sequence in the FG loop (xYxGPxxYGxxEx), which will be investigated in a future study.
これらのクローンは、FGループ(xYxGPxxYGxxEx)で保存されたシーケンスを示しました。これは、今後の研究で調査されます。
Monobody clones 4 and 9 shared another epitope that was also likely located in the ACE2-binding surface (Figs. 3B and 3D).
モノボディクローン4と9は、ACE2結合表面にある可能性が高い別のエピトープを共有していました(図3Bと3D)。
No conserved sequence was detected in the BC loop or the FG loop of these clones.
これらのクローンのBCループまたはFGループでは、保存された配列は検出されませんでした。
Therefore, we identified 16 pairs that can be used in further sandwich ELISA.
したがって、さらにサンドイッチELISAで使用できる16ペアを特定しました。
Among them, we tested clone 10 as a capturing monobody and clone 12 as a detecting monobody, because the pair gave one of the highest signal-to-noise ratios in the sandwich ELISA experiment (Fig. 3D).
このうち、クローン10を捕捉モノボディとして、クローン12を検出モノボディとしてテストしました。これは、ペアがサンドイッチELISA実験で最も高いS / N比を示したためです(図3D)。
The results of sandwich ELISA showed that a combination of monobodies could specifically detect the SARS-CoV-2 spike protein trimer and the S1 subunit (Fig. 3E).
サンドイッチELISAの結果は、モノボディの組み合わせが、SARS-CoV-2スパイクタンパク質三量体とS1サブユニットを特異的に検出できることを示しました(図3E)。
Although the sensitivity of this system was not sufficient to detect the target in actual samples (103 particles of SARS-CoV2 in a 100 μL solution, corresponding to 1 fM spike protein), its combination with other detection methods, such as digital ELISA or loop-mediated isothermal amplification, may help to overcome the problem of the detection limit in a future study.
このシステムの感度は実際のサンプルでターゲットを検出するのに十分ではありませんでしたが(1μMスパイクタンパク質に対応する100μL溶液中のSARS-CoV2の103粒子)、デジタルELISAやループを介した等温増幅などの他の検出方法との組み合わせは、将来の研究における検出限界の問題の克服に役立つ可能性があります。
Capture of SARS-CoV-2 virions using monobodies from a cultivated sample
培養されたサンプルからのモノボディを使用したSARS-CoV-2ビリオンの捕捉
Next, we studied the binding of monobodies to SARS-CoV-2.
次に、SARS-CoV-2へのモノボディの結合を研究しました。
Each monobody-biotin was incubated with SARS-CoV-2 particles in phosphate-buffered saline (PBS) and the viral particles bound to the monobodies were collected on the magnetic beads.
各モノボディビオチンをリン酸緩衝生理食塩水(PBS)でSARS-CoV-2粒子と培養し、モノボディに結合したウイルス粒子を磁気ビーズに集めました。
An RT-qPCR assay of the SARS-CoV-2 RNA extract in the supernatants and the bead eluents showed that a large proportion of the SARS-CoV-2 particles was detected in the bead eluents for all monobodies, with the exception of the WT monobody (Fig. 4A), proving the binding activity of all selected monobodies toward SARS-CoV-2 particles.
上清およびビーズ溶出液中のSARS-CoV-2 RNA抽出物のRT-qPCRアッセイは、すべてのモノボディのビーズ溶出液でSARS-CoV-2粒子の大きな割合が検出されたことを示しました。ただしWTモノボディは例外です(図4A)。このことは、SARS-CoV-2粒子に対する選択されたすべてのモノボディの結合活性を証明します。
Based on the experiment described above, we realized that this pull-down method also would be useful for enhancing the detection limit of SARS-CoV-2.
上記の実験に基づいて、このプルダウン方法はSARS-CoV-2の検出限界を高めるためにも役立つことがわかりました。
To test this idea, SARSCoV-2 particles were suspended in a PBS solution at various concentrations (0.1–10,000 particles/μL), and the number of particles was analyzed by pull-down RT-qPCR using monobody clone 4 or by a traditional RT-qPCR.
このアイデアをテストするために、SARSCoV-2粒子をさまざまな濃度(0.1〜10,000粒子/μL)のPBS溶液に懸濁しました。粒子の数は、モノボディクローン4を使用したプルダウンRT-qPCRまたは従来のRT-qPCRによって分析されました。
The result showed a wide linear dynamic range of this method (Fig. 4B).
結果は、この方法の広い線形ダイナミックレンジを示しました(図4B)。
The detection limit indeed was increased from 1 to 0.1 particles/μL.
実際、検出限界は1から0.1粒子/μLに増加しました。
Monobody capture of SARS-CoV-2 virions from nasal swab samples of patients
患者の鼻腔スワブサンプルからのSARS-CoV-2ビリオンのモノボディキャプチャ
We then performed a pull-down of SARS-CoV-2 particles from nasal swab samples of patients (Fig. 4C).
次に、患者の鼻腔スワブサンプルからSARS-CoV-2粒子のプルダウンを実行しました(図4C)。
Of note, the actual patient samples (sometimes in the form of highly viscous liquids) were very different from cultured samples, primarily because they contain many unknown components.
注目に値するのは、実際の患者のサンプル(高粘度の液体の場合もある)は、主に未知の成分が多く含まれているため、培養されたサンプルとは非常に異なっていたということです。
We tested 16 samples, and found that the monobody captured SARS-CoV-2 even in these nasal swab samples.
16個のサンプルをテストしたところ、これらの鼻腔スワブサンプルでも、モノボディがSARS-CoV-2を捕捉していることがわかりました。
To our knowledge, this is the first example of an antibody or an ALP to capture SARS-CoV-2 from patient nasal swab samples.
私たちの知る限り、これは患者の鼻腔スワブサンプルからSARS-CoV-2を捕捉するための抗体またはALPの最初の例です。
Interestingly, for samples #8 , #15 , and #16 , the capture rates were significantly lower than the remaining cases.
興味深いことに、サンプル#8、#15、および#16では、キャプチャ率が他のケースよりも大幅に低くなっています。
We reasoned that this might be because the monobody binding site was hidden by neutralizing antibodies developed in the patients.
これは、モノボディ結合部位が患者で開発された中和抗体によって隠されていたためと考えられます。
Finally, we performed a pull-down direct RT-qPCR for nasal swab samples (400 μL) of patients (Fig. 4D).
最後に、患者の鼻腔スワブサンプル(400μL)に対してプルダウン直接RT-qPCRを実行しました(図4D)。
We tested four samples this way and found that the monobody captured SARS-CoV-2 even in higher-volume nasal swab samples.
この方法で4つのサンプルをテストしたところ、大量の鼻腔スワブサンプルでも、モノボディがSARS-CoV-2を捕捉することがわかりました。
The number of RNA genomes were increased 25–77 times after the pull-down technique was employed.
RNAゲノムの数は、プルダウンテクニックが採用された後、25〜77倍に増加しました。
Moreover, RNA genome in the nasal swab sample #4 was detected only by the pull-down direct RT-qPCR but not by traditional direct RTqPCR, suggesting the increased sensitivity of the pull-down direct RT-qPCR assay.
さらに、鼻腔スワブサンプル#4のRNAゲノムはプルダウンダイレクトRT-qPCRでのみ検出され、従来のダイレクトRTqPCRでは検出されなかったため、プルダウンダイレクトRT-qPCRアッセイの感度が向上しました。
Binding activity of monobody tandem dimers
モノボディタンデムダイマーの結合活性
A dimeric form of a protein binder usually showed higher affinity than the monomeric one;
タンパク質結合剤の二量体は通常、単量体結合体よりも高い親和性を示しました。
therefore, we next synthesized the tandem dimers of a monobody.
したがって、次に、モノボディのタンデムダイマーを合成しました。
We chose clone 4, 6b, and 12b to synthesize the corresponding tandem dimers (TD4, TD6b, and TD12b) because these clones showed the highest affinity in each group defined by the binding competition assay (Fig. 3D).
対応するタンデムダイマー(TD4、TD6b、およびTD12b)を合成するために、クローン4、6b、および12bを選択しました。これらのクローンは、結合競合アッセイによって定義された各グループで最も高い親和性を示したためです(図3D)。
As expected, the affinities of these tandem dimers were drastically improved (Fig. 5A, Table 1):
予想通り、これらのタンデムダイマーの親和性は大幅に改善されました(図5A、表1)。
the KD displayed less than 1 pM because of the immeasurable koff value (10−7 /s).
測定できないkoff値(10-7 / s)のため、KDは1 pM未満で表示されました。
To understand why the dimerization of monobodies drastically improved the affinity, we first immobilized decreased amounts of S1 protein on the sensor chip and analyzed monobody dissociation.
モノボディの二量体化によって親和性が大幅に改善された理由を理解するために、まずセンサーチップ上に減少した量のS1タンパク質を固定化し、モノボディの解離を分析しました。
The results showed that the monobody dimers still dissociated very slowly (Fig. S7A).
結果は、モノボディ二量体が依然として非常にゆっくりと解離することを示しました(図S7A)。
Next, when the monobodies were immobilized on the sensor chip and the S1 protein was used as an analyte, we observed a similar KD value for Clone 6b (Fig, 2 vs. Fig. S7B, Table 1) and no improvement on the KD values for the tandem dimers (Fig, 2 vs. Figure 5B, Table 1).
次に、モノボディがセンサーチップに固定化され、S1タンパク質が分析物として使用された場合、クローン6bに対して同様のKD値が観察されました(図2と図S7B、表1)。タンデムダイマーのKD値は改善されていません(図2と図5B、表1の比較)。
These results suggest that the tandem connection of monobodies does not improve the affinity, but bringing up the bridging effect, i.e., an avidity effect.
これらの結果は、モノボディのタンデム接続が親和性を改善するのではなく、ブリッジング効果、つまりアビディティ効果を生み出すことを示唆しています。
※アビディティ=IgMなどの多価抗体が抗原に結合する時の結合力の総和
Monobodies exhibited a high neutralizing activity against SARS-CoV-2 infection
モノボディは、SARS-CoV-2感染に対して高い中和活性を示しました
The monobody clone 6b showed high affinity against the S1 subunit of the SARS-CoV-2 spike protein and inhibited the interaction between ACE2 and the S1 subunit (Fig. 3C).
モノボディクローン6bは、SARS-CoV-2スパイクタンパク質のS1サブユニットに対して高い親和性を示し、ACE2とS1サブユニット間の相互作用を阻害しました(図3C)。
These findings led us to analyze the neutralizing activity of monobody clone 6b and TD6b against SARS-CoV-2 infection.
これらの調査結果から、SARS-CoV-2感染に対するモノボディクローン6bおよびTD6bの中和活性を分析することができました。
We added 4-fold serially diluted monobody solutions to VeroE6/TMPRSS2 cells(34) in 96-well culture plates, followed by infection with SARS-CoV-2 (105 TCID50/mL) for 1 hour at 37°C.
4倍連続希釈モノボディ溶液を96ウェル培養プレートのVeroE6 / TMPRSS2細胞(34)に追加し、37℃で1時間SARS-CoV-2(105 TCID50 / mL)に感染させました。
After 36 hours of incubation with fresh medium, SARS-CoV-2 mRNA amounts in the supernatant were measured by qRT-PCR.
新しい培地との36時間の培養後、上清中のSARS-CoV-2 mRNA量をqRT-PCRで測定しました。
The monobody clone 6b showed a very low half maximal inhibitory concentration (IC50 = 0.5 nM, Fig. 5C), even in the monomeric form.
モノボディクローン6bは、単量体型であっても、最大阻害濃度の半分(IC50 = 0.5 nM、図5C)を示しました。
Its neutralizing activity was increased in the dimeric form (IC50 = 0.4 nM), although the data obtained for 390 pM TD6b fluctuated, probably because of the high sensitivity of VeroE6/TMPRSS2 cells.
中和活性は二量体の形で増加しました(IC50 = 0.4 nM)。ただし、390 pM TD6bで得られたデータは変動しましたが、おそらくVeroE6 / TMPRSS2セルの感度が高いためです。
More importantly, these IC50 values were comparable to those of the high-affinity human neutralizing antibodies reported recently, indicating the usefulness of TRAP display for the selection of highly active ALPs.
さらに重要なことに、これらのIC50値は最近報告された高親和性ヒト中和抗体に匹敵し、非常にアクティブなALPを選択するためのTRAP表示の有用性を示しています。
Discussion
Here, we developed a high-speed in vitro selection method to obtain ALPs against various targets.
ここでは、さまざまなターゲットに対してALPを取得するための高速in vitro選択方法を開発しました。
We demonstrated a constant development of high-affinity binders (KD values in the low- or sub-nM range) against EGFR1, HER2, and the SARSCoV-2 spike protein.
EGFR1、HER2、およびSARSCoV-2スパイクタンパク質に対する高親和性バインダー(低またはサブnM範囲のKD値)の一定の発展を実証しました。
Furthermore, our selected monobodies recognized a recombinant spike protein as well as SARS-CoV2 particles, demonstrating applicability for a sandwich ELISA for a rapid antigen test.
さらに、選択したモノボディは組換えスパイクタンパク質とSARS-CoV2粒子を認識し、迅速な抗原検査のためのサンドイッチELISAへの適用性を実証しました。
Using the monobody before performing pull-down RT-qPCR also increased the detection limit of SARS-CoV-2.
プルダウンRT-qPCRを実行する前にモノボディを使用すると、SARS-CoV-2の検出限界も増加しました。
More importantly, the monobody captured SARS-CoV-2 in nasal swab samples of COVID-19 patients.
さらに重要なのは、モノボディがCOVID-19患者の鼻腔スワブサンプルでSARS-CoV-2を捕捉したことです。
Actual patient samples are very different from cultured samples because they consist of inhomogeneous liquid and contain many unknown components, including various proteases, mucins, and neutralization antibodies.
実際の患者のサンプルは、不均質な液体で構成されており、さまざまなプロテアーゼ、ムチン、中和抗体などの未知の成分を多く含んでいるため、培養サンプルとは大きく異なります。
We believe that the monobodies procured in our study will soon be useful to develop effective diagnostic tools and that such tools will contribute to the worldwide effort to overcome the COVID-19 pandemic.
私たちの調査で入手したモノボディはすぐに効果的な診断ツールを開発するのに役立ち、そのようなツールはCOVID-19パンデミックを克服するための世界的な取り組みに貢献すると考えています。
The monobody also had a very low IC50 value against SARS-CoV-2 infection, which was comparable to those of the antibodies that were reported recently as being highly active for neutralizing SARS-CoV-2.
モノボディはまた、SARS-CoV-2感染に対して非常に低いIC50値を示しました。これは、SARS-CoV-2を中和する活性が高いと最近報告された抗体に匹敵しました。
Because the backbone of the monobody was the 10th type III domain of human fibronectin, it might be useful as a neutralizing ALP against SARS-CoV-2 infection.
モノボディのバックボーンは、ヒトフィブロネクチンの10番目のタイプIIIドメインであったため、SARS-CoV-2感染に対するALPの中和として役立つ可能性があります。
※フィブロネクチン=巨大な糖タンパク質で、細胞接着分子。
We received the SARS-CoV-2 spike protein S1 subunit on April 7, 2020 as a target of the TRAP display selection and, using the methods described herein, obtained high-affinity monobody sequences on April 10, 2020.
2020年4月7日、TRAPディスプレイ選択のターゲットとしてSARS-CoV-2スパイクプロテインS1サブユニットを受け取り、2020年4月10日に、ここで説明する方法を使用して高親和性モノボディシーケンスを取得しました。
The high- speed feature of the improved TRAP display is therefore useful for a rapid response to subspecies of SARS-CoV-2 in addition to potential new viruses causing future pandemics.
したがって、改良されたTRAP表示の高速機能は、将来のパンデミックを引き起こす可能性のある新しいウイルスに加えて、SARS-CoV-2の亜種への迅速な応答に役立ちます。
Materials and Methods
Materials
材料
The oligonucleotides were purchased either Fasmac Co., Ltd. (Japan) or Nippon Bio Service (Japan).
オリゴヌクレオチドは、Fasmac Co.、Ltd.(日本)またはNippon Bio Service(日本)のいずれかで購入しました。
The primer/probe set for RT-qPCR and positive control RNA for N2 were purchased from Applied Biosystems, Inc. (NY, USA).
RT-qPCRのプライマー/プローブセットとN2のポジティブコントロールRNAは、Applied Biosystems、Inc.(NY、USA)から購入しました。
The synthetic DNAs were acquired from GenScript (NJ, USA).
合成DNAはGenScript(NJ、USA)から取得しました。
The sequences of the primers and synthetic DNAs are listed in Data file S1.
プライマーと合成DNAのシーケンスは、データファイルS1にリストされています。
SARS-CoV-2 protein active trimer (SPN-C52H8), human ACE2 protein (AC2-H5257), biotinylated 2019-nCoV S1 subunit-Avitag (S1N-C82E8), biotinylated 2019-nCoV S protein RBD-Avitag (SPD-C82E9), SARS-CoV-2 (COVID-19) S1 subunit (S1N-C52H4), and SARS S1 subunit (S1N-S52H5) were all purchased from ACROBiosystems (DE, USA).
SARS-CoV-2タンパク質活性三量体(SPN-C52H8)、ヒトACE2タンパク質(AC2-H5257)、ビオチン化2019-nCoV S1サブユニット-Avitag(S1N-C82E8)、ビオチン化2019-nCoV Sタンパク質RBD-Avitag(SPD-C82E9 )、SARS-CoV-2(COVID-19)S1サブユニット(S1N-C52H4)、SARS S1サブユニット(S1N-S52H5)はすべてACROBiosystems(DE、USA)から購入したものです。
EGF receptor/Fc chimera and ErbB2/Fc chimera were obtained from R&D systems (MN, USA).
EGF受容体/ FcキメラおよびErbB2 / Fcキメラは、R&Dシステム(MN、USA)から入手しました。
Creatine kinase, creatine phosphate, and E. coli tRNAs were purchased from Roche Diagnostics (Japan).
クレアチンキナーゼ、クレアチンリン酸塩、およびE. coli tRNAは、Roche Diagnostics(日本)から購入しました。
The restriction enzymes were obtained from New England Biolabs (MA, USA).
制限酵素は、New England Biolabs(MA、USA)から入手しました。
Preparation of monobody mRNA libraries for selection against SARS-CoV-2 targets
SARS-CoV-2ターゲットに対する選択のためのモノボディmRNAライブラリの準備
To prepare an A-fragment DNA of the monobody library, FN3F0.F83 (1μM), FN3F1-2.F29(P) (1μM), and the FN3FF1coR8.F73 (0.5 μM) or FN3FF1coR10.F79 (0.5 μM) were ligated by T4 DNA ligase (75 μL in total, 75 pmol for each oligonucleotide) with an assistance of Fn3an1.R20(3NH2) (2 μM), and Fn3an2-1.R20(3NH2) (2 μM).
モノボディライブラリのAフラグメントDNAを準備するには、FN3F0.F83(1μM)、FN3F1-2.F29(P)(1μM)、およびFN3FF1coR8.F73(0.5μM)またはFN3FF1coR10.F79(0.5μM)は、Fn3an1.R20およびFn3an2-1.R20(3NH2) (2 μM)の支援を得てT4 DNAリガーゼ(合計75μL、各オリゴヌクレオチドで75 pmol)でライゲーションされました。
As a codons for the randomized residues, we used a codon mix with the following ratios:
ランダム化された残基のコドンとして、次の比率のコドンミックスを使用しました。
20% Tyr, 10% Ser, 15% Gly, 10% Trp, and 3% each of all the other amino acids except for Cys, which is similar to the original cocktail (30% Tyr, 15% Ser, 10% Gly, 5% Phe, 5% Trp, and 2.5% each of all the other amino acids except for Cys).
20%Tyr、10%Ser、15%Gly、10%Trp、およびCysを除く他のすべてのアミノ酸のそれぞれ3%。これは、元のカクテル(Tyr 30%、Ser 15%、Gly 10%、Phe 5%、Trp 5%、Cysを除く他のすべてのアミノ酸それぞれ2.5%)に似ています。
After the ligation, the entire mixture was added to the reaction mixture (10 mM Tris-HCl pH 8.4, 100 mM KCl, 0.1% (v/v) Triton X-100, 2% (v/v) DMSO, 2 mM MgSO4, 0.2 mM each dNTP, 0.375 μM T7SD8M2.F44, 0.375 μM FN3BsaI.R40, and 2 nM of Pfu-S DNA polymerase) and the amplified by PCR (15 mL in total, seven cycles of PCR).
ライゲーション後、混合物全体を反応混合物に加え、PCRで増幅しました(合計15 mL、7サイクルのPCR)。
反応混合物:10 mM Tris-HCl pH 8.4・100 mM KCl, 0.1% (v/v) Triton X-100・2% (v/v) DMSO・2 mMのMgSO4、各0.2 mMのdNTP・0.375 μM T7SD8M2.F44・0.375 μM FN3BsaI.R40・2 nMのPfu-S DNAポリメラーゼ
B-fragment DNA was prepared by the same procedure using FN3FF2co.F72(p), FN3F3coR10.F70(p), FN3F3coR12.F76(p), Fn3an3.R20(3NH2) for ligation, and FN3BsaI.F33, FN3Pri2.R44 for amplification.
BフラグメントDNAは、ライゲーションにはFN3FF2co.F72(p)・N3F3coR12.F76(p)・ Fn3an3.R20(3NH2)を、増幅にはN3BsaI.F33・FN3Pri2.R44を使用して、同じ手順で調製しました。
The amplified A-fragment DNA and B-fragment DNA were purified by phenol/chloroform extraction and isopropanol precipitation.
増幅されたAフラグメントDNAとBフラグメントDNAは、フェノール/クロロホルム抽出とイソプロパノール沈殿によって精製されました。
One end of each DNA product was digested with BsaI (New England Biolabs, MA, USA) as per the manufacturer’s protocol, and the DNA products were then purified by phenol/chloroform extraction and isopropanol precipitation.
各DNA産物の一方の端をメーカーのプロトコルに従ってBsaI(New England Biolabs、MA、USA)で消化し、フェノール/クロロホルム抽出とイソプロパノール沈殿によりDNA産物を精製しました。
The products were ligated to each other (1 μM, 200 μL) to synthesize full-length DNA products, and they were amplified using T7SD8M2.F44, G5S-4Gan21-3.R42, and Pfu-S DNA polymerase (60 mL in total, four cycles of PCR).
製品を互いにライゲーション(1μM、200μL)して、完全長のDNA製品を合成し、T7SD8M2.F44, G5S-4Gan21-3.R42およびPfu-S DNAポリメラーゼを使用して増幅しました。(合計60 mL、4サイクルのPCR)
The products were purified through phenol/chloroform extraction and isopropanol precipitation.
製品は、フェノール/クロロホルム抽出とイソプロパノール沈殿によって精製されました。
The DNA template was transcribed by in vitro run-off transcription, and the mRNA was purified by isopropanol precipitation followed by PAGE purification.
DNAテンプレートはin vitroランオフトランスクリプションによって転写され、mRNAはイソプロパノール沈殿とその後のPAGE精製によって精製されました。
The mRNA/HEX-mPuL was prepared by a similar procedure described above.
mRNA / HEX-mPuLは、上記と同様の手順で調製しました。
The resulting complex was directly used in the first-round of selection.
得られた複合体は、最初の選択ラウンドで直接使用されました。
In vitro selection of monobodies against SARS-CoV-2 Spike protein S1 subunit and RBD of the S1 subunit by the improved TRAP display
改善されたTRAP表示による、SARS-CoV-2スパイクタンパク質S1サブユニットおよびS1サブユニットのRBDに対するモノボディのin vitro選択
For first-round selection, 1 μM mRNA/ Pu-OMe-linker was added to a reconstituted translation system, and the reaction mixture (500 μL) was incubated at 37°C for 30 min.
1回目の選択では、1μMmRNA / Pu-OMe-linkerを再構成した翻訳システムに添加し、反応混合物(500μL)を37℃で30分間培養しました。
After the reaction, 41.7 μL of 200 mM EDTA (pH 8.0) was added to the translation mixture.
反応後、41.7μLの200 mM EDTA(pH 8.0)を翻訳混合物に添加しました。
A reverse transcription buffer (41.1 μL of 0.78 M Tris-HCl pH 8.4, 1.16 M KCl, 0.37 M MgCl2 and 0.08 M DTT), 5 mM dNTPs (66.7 μL), 100 μM FN3S.R29 (10 μL), and 28.7 μM HMLV (27.5 μL) were added to the translation mixture, and the resulting solution was incubated at 42°C for 15 min.
逆転写バッファー(41.1μLの0.78 M Tris-HCl pH 8.4、1.16 M KCl、0.37 M MgCl2および0.08 M DTT)、5 mM dNTP (66.7 μL)、100μM FN3S.R29 (10 μL)、および28.7μMHMLV (27.5 μL)を翻訳混合物に添加し、得られた溶液を42℃で15分間培養しました。
The buffer was exchanged to HBST bufer using Zeba Spin Desalting Columns.
Zebaスピン脱塩カラムを使用して、バッファーをBSTバッファーに交換しました。
In order to remove the bead binders, the resulting solution was mixed with Dynabeads M-280/M270 streptavidin (1:1) (Thermo Fisher cientific) at 25°C for 10 min.
ビーズバインダーを除去するために、得られた溶液をDynabeads M-280 / M270ストレプトアビジン(1:1)(Thermo Fisher cientific)と25℃で10分間混合しました。
The supernatant was mixed with 4.8 μL of 7.3 μM biotinylated 2019-nCoV S1 subunit-Avitag and 2.1 μL of 16.5 μM biotinylated 2019-nCoV S protein RBD-Avitag, and then incubated at 25°C for 5 min.
上清を4.8μLの7.3μMビオチン化2019-nCoV S1サブユニット-Avitagおよび2.1μLの16.5μMビオチン化2019-nCoV Sタンパク質RBD-Avitagと混合し、25°Cで5分間培養しました。
After recovering the target proteins with Dynabeads M-270 streptavidin, the resulting beads were washed with HBST buffer thrice, and PCR premix (1 mL) was added.
Dynabeads M-270ストレプトアビジンで標的タンパク質を回収した後、得られたビーズをHBSTバッファーで3回洗浄し、PCRプレミックス(1 mL)を加えました。
The beads were heated at 95°C for 5 min, and the amount of the eluted cDNAs was quantified by SYBR greenbased quantitative PCR using T7SD8M2.F44 and FN3Lip.R20 as primers.
ビーズを95℃で5分間加熱し、T7SD8M2.F44とFN3Lip.R20をプライマーとして使用して、SYBRグリーンベースの定量的PCRによって溶出されたcDNAの量を定量しました。
The eluted cDNAs were PCR-amplified using T7SD8M2.F44, G5S-4Gan21-3.R42, and Pfu-S DNA polymerase, and was purified by phenol/chloroform extraction and isopropanol precipitation.
溶出されたcDNAは、T7SD8M2.F44、G5S-4Gan21-3.R42、およびPfu-S DNAポリメラーゼを使用してPCR増幅され、フェノール/クロロホルム抽出およびイソプロパノール沈殿によって精製されました。
The post-translation and post-reverse transcription reaction mixtures were analyzed by using the same procedure as described above.
上記と同じ手順を使用して、翻訳後および逆転写後の反応混合物を分析しました。
For the second-round selection, the resulting DNA (about 3–10 nM in final concentration) was added to the TRAP system and the reaction mixture (40 μL) was incubated at 37°C for 30 min.
2回目の選択では、得られたDNA(最終濃度で約3〜10 nM)をTRAPシステムに加え、反応混合物(40μL)を37℃で30分間培養しました。
After the reaction, 8 μL of 100 mM EDTA (pH 8.0) was added to the translation mixture.
反応後、8μLの100 mM EDTA(pH 8.0)を翻訳混合物に加えました。
Reverse transcription mixture (24 μL; 150 mM Tris-HCl pH 8.4, 225 mM KCl, 75 mM MgCl2 and 16 mM DTT, 1.5 mM dNTPs, 7.5 μM primer, and 3.4 μM HMLV) were added to the translation mixture, and the resulting solution was incubated at 42°C for 15 min.
Reverse transcription mixture were added to the translation mixture, and the resulting solution was incubated at 42°C for 15 min.
逆転写混合物(24μL、150 mM Tris-HCl pH 8.4、225 mM KCl、75 mM MgCl2および16 mM DTT、1.5 mM dNTP、7.5μMプライマー、および3.4μMHMLV)を翻訳混合物に加え、得られた溶液を42°Cで15分間培養しました。
The buffer was exchanged to HBST buffer using Zeba Spin Desalting Columns.
Zebaスピン脱塩カラムを使用して、バッファーをHBSTバッファーに交換しました。
To remove bead binders, the resulting solution was mixed thrice with M270/M280 magnetic beads (1:1) at 25°C for 20 min.
ビーズバインダーを除去するために、得られた溶液をM270 / M280磁気ビーズ(1:1)と25℃で20分間3回混合しました。
Each target (20 nM final) was added to the half portion of the supernatant.
各ターゲット(最終20 nM)を上澄みの半分に加えました。
After removing the supernatant, the beads were washed with HBST buffer thrice, and the PCR premix was added to the beads.
上清を除去した後、ビーズをHBSTバッファーで3回洗浄し、PCRプレミックスをビーズに追加しました。
Quantitation of cDNA and amplification and purification of DNA were performed by the same procedure as described for the firstround selection.
cDNAの定量とDNAの増幅と精製は、最初の選択と同じ手順で行いました。
For the third- to sixth-round selection, the procedure was performed similarly as that of the second-round, except for the volume of the reaction mixtures (5 μL), the target concentration (2 nM in fourth-round to sixth-round), and bead washing conditions (an extensive wash in the sixth-round selection against S1 subunit).
3回目から6回目までの選択では、反応混合物の容量(5μL)、目標濃度(4ラウンドから6ラウンドで2 nM)、およびビーズの洗浄条件(S1サブユニットに対する第6ラウンドの選択での広範な洗浄)を除いて、手順は2回目と同じように実行されました。
After the sixth-round selection, the sequences of the recovered DNAs were analyzed using an Ion Torrent instrument (Thermo Fisher Scientific).
6回目の選択の後、回収されたDNAのシーケンスは、Ion Torrent機器(Thermo Fisher Scientific)を使用して分析されました。
Affinity measurement of selected nanobodies and monobodies
選択したナノボディとモノボディの親和性測定
The affinity measurement was performed against the EGF receptor/Fc chimera or the ErbB2/Fc chimera immobilized on an anti-Human IgG Fc Capture biosensor (ForteBio), biotinylated 2019-nCoV S1 subunit-Avitag or biotinylated monobodies immobilized on a streptavidin biosensor (ForteBio), using Octet system (ForteBio, CA, USA), as described in the manufacturer’s instructions.
親和性測定は次のものに対し行われました。
・抗ヒトIgG Fcキャプチャバイオセンサー(ForteBio)に固定化された、EGF受容体/ FcキメラまたはErbB2 / Fcキメラ。
・ストレプトアビジンバイオセンサー(ForteBio)に固定化された、ビオチン化2019-nCoV S1サブユニット-Avitagまたはビオチン化モノボディ
メーカーの指示に従って、Octetシステム(ForteBio、CA、USA)を使用します。
The binding assay was performed at 30°C in the buffer D.
結合アッセイは、バッファーD中で30℃で行った。
Each step in the binding assay was as follows:
バインディングアッセイの各ステップは次のとおりです。
equilibration for 150 s, association for 900 s, and dissociation for 900 s (EGF and HER2);
150秒間の平衡、900秒間の結合、900秒間の解離(EGFとHER2)
equilibration for 150 s, association for 600 s, and dissociation for 600 s (S1 subunit).
150秒間の平衡、600秒間の結合、600秒間の解離(S1サブユニット)。
Buffer D’ [50 mM Hepes-KOH pH 7.5, 300 mM NaCl, 0.1% (v/v) Tween 20, and 1% (w//v) PEG6000] was used when the S1 subunit was an analyte.
バッファーD ’[50 mM Hepes-KOH pH 7.5、300 mM NaCl、0.1%(v / v)Tween 20、および1%(w // v)PEG6000]は、S1サブユニットが分析物である場合に使用されました。
Binding assay of monobodies against SARS-CoV-2 S1 or S1 RBD proteins
SARS-CoV-2 S1またはS1 RBDタンパク質に対するモノボディの結合アッセイ
The ELISA microplate with 96 wells was coated with 100 μL of 100 nM monobody in HBS2 buffer (25 mM Hepes-K pH 7.5, 150 mM NaCl) overnight.
96ウェル付きのELISAマイクロプレートを、HBS2バッファー(25 mM Hepes-K pH 7.5、150 mM NaCl)中の100 nLモノボディ100μLで一晩コーティングしました。
The microplate was washed once with HBST2 buffer [HBS2 with 0.05% (v/v) Tween 20)], blocked with 1% (w/v) bovine serum albumin (BSA) in HBS2 buffer at room temperature for one hour, and then was washed once with HBST2 buffer.
マイクロプレートをHBST2バッファー[0.05%(v / v)Tween 20)を含むHBS2で1回洗浄し、HBS2バッファー中の1%(w / v)ウシ血清アルブミン(BSA)で室温で1時間ブロックした後、HBST2バッファーで1回洗浄した。
S1–HRP or RBD–HRP was prepared by mixing biotinylated SARS-CoV-2 spike protein S1 subunit (S1-biotin) or the receptor binding domain (RBDbiotin) with the same amount of streptavidin-HRP (Stv-HRP) in HBST2BP buffer [HBST2 with 0.1% (w/v) BSA and 0.1% (w/v) PEG6000], and was added to the microplate with a final concentration of 10 nM.
S1-HRPまたはRBD-HRPは、ビオチン化SARS-CoV-2スパイクタンパク質S1サブユニット(S1-ビオチン)または受容体結合ドメイン(RBDビオチン)をHBST2BPバッファー[0.1%(w / v)BSAおよび0.1%(w / v)PEG6000を含むHBST2]内の同量のストレプトアビジン-HRP(Stv-HRP)と混合することで準備され、そして、最終濃度10 nMでマイクロプレートに加えられました。
The microplate was incubated at room temperature for one hour.
マイクロプレートを室温で1時間培養しました。
The microplate was washed five times with HBST2 and 200 μL of SuperSignal ELISA Pico was added to each well.
マイクロプレートをHBST2で5回洗浄し、200μLのSuperSignal ELISA Picoを各ウェルに加えました。
The chemiluminescence was measured immediately using the SpectraMax M5 plate reader (Molecular Devices).
化学発光は、SpectraMax M5プレートリーダー(Molecular Devices)を使用して直ちに測定しました。
Pull-down of S proteins with monobody-immobilized beads
モノボディ固定化ビーズによるSタンパク質のプルダウン
Biotinylated monobody (1 μM, 3.3 μL) in HBST2BP buffer was mixed with Dynabeads M-280 streptavidin (10 mg/mL, 6.6 μL, pre-washed by HBST buffer) at room temperature for 5 min.
HBST2BPバッファー内のビオチン化モノボディ(1μM、3.3μL)をDynabeads M-280ストレプトアビジン(10 mg / mL、6.6μL、HBSTバッファーで事前洗浄)と室温で5分間混合しました。
After removing the supernatant, the beads were washed twice with HBST buffer, and 2.5 μL of 100 nM each analyte (SARS-CoV-2 spike protein S1 subunit, SARS-CoV spike protein S1 subunit, SARS-CoV spike protein trimer) in buffer D was added to a one-third fraction of the beads.
上澄みを取り除いた後、ビーズをHBSTバッファーで2回洗浄し、バッファーD中の各100 nM分析物2.5μLをビーズの3分の1のフラクションに加えました。
各分析物:SARS-CoV-2スパイクタンパク質S1サブユニット、SARS-CoVスパイクタンパク質S1サブユニット、SARS-CoVスパイクタンパク質三量体
After mixing at room temperature for 30 min, the supernatant was collected.
室温で30分間混合した後、上澄みを回収しました。
The beads were washed twice with HBST buffer.
ビーズをHBST緩衝液で2回洗浄した。
The supernatant and heat-elution from the beads were loaded on SDS-PAGE gel and was stained using Sypro Ruby.
ビーズの上清と熱溶出液をSDS-PAGEゲルにロードし、Sypro Rubyを使用して染色しました。
Inhibition of ACE2 / CoV-2 S1 subunit interaction by monobodies
モノボディによるACE2 / CoV-2 S1サブユニット相互作用の阻害
ELISA was performed using a microplate with 96 wells, coated with 100 μL of 2 nM SARS-CoV-2 Spike protein S1 subunit in HBS2 buffer overnight.
ELISAは、96ウェルのマイクロプレートを使用して、HBS2バッファー中の2 nM SARS-CoV-2 Spikeタンパク質S1サブユニット100μLで一晩コーティングして行いました。
The microplate was washed once with HBST2 buffer, blocked with 1% (w/v) BSA in HBS2 buffer at room temperature for one hour, and was then washed once with HBST2 buffer.
マイクロプレートをHBST2バッファーで1回洗浄し、HBS2バッファー中の1%(w / v)BSAで室温で1時間ブロックした後、HBST2バッファーで1回洗浄しました。
Biotinylated angiotensinconverting enzyme 2 (ACE2-biotin) complexed with same amount of Stv-HRP in HBST2BP buffer was added to a final concentration of 1 nM together with 100 nM or 10 nM monobody.
HBS 2BPバッファー中の同量のStv-HRPと複合体を形成したビオチン化アンジオテンシン変換酵素2(ACE2-ビオチン)を、100 nMまたは10 nMモノボディと共に1 nMの最終濃度になるように添加しました。
Alternatively, a monobody (10 nM in total) was added to the ACE2–HRP-preincubated S1-immobilized microplate.
または、ACE2–HRPでプレインキュベートしたS1固定化マイクロプレートにモノボディ(合計10 nM)を追加しました。
The microplate was incubated for one hour at 37°C.
マイクロプレートを37℃で1時間インキュベートした。
The plate was then washed five times with HBST2 buffer.
次に、プレートをHBST2バッファーで5回洗浄しました。
After addition of SuperSignal ELISA Pico (200 μL) to each well, the chemiluminescence was recorded immediately using a SpectraMax M5 plate reader.
SuperSignal ELISA Pico(200μL)を各ウェルに添加した後、SpectraMax M5プレートリーダーを使用して化学発光を直ちに記録しました。
Sandwich ELISA using monobodies
モノボディを使用したサンドイッチELISA
The sandwich ELISA was performed using a microplate with 384 wells, which was coated with 25 μL of 100 nM of the capturing monobodies (9 clones plus WT) in HBS2 buffer overnight.
サンドイッチELISAは、HBS2バッファー中の100 nMの捕捉モノボディ(9クローン+ WT)25μLで一晩コーティングされた384ウェルのマイクロプレートを使用して実行されました。
The microplate was washed once with HBST2 buffer, blocked with 1% (w/v) BSA in HBS2 buffer at room temperature for one hour, and was then washed once with HBST2 buffer.
マイクロプレートをHBST2バッファーで1回洗浄し、HBS2バッファー中の1%(w / v)BSAで室温で1時間ブロックした後、HBST2バッファーで1回洗浄しました。
SARS-CoV-2 S1 subunit (10 nM) in HBST2BP buffer was added.
HBST2BPバッファー内のSARS-CoV-2 S1サブユニット(10 nM)が追加されました。
The microplate was incubated at room temperature for one hour, and was subsequently washed once with HBST2 buffer.
マイクロプレートを室温で1時間インキュベートした後、HBST2バッファーで1回洗浄しました。
Biotinylated detecting monobody (9 clones plus WT) complexed with same amount of Stv-HRP in HBST2BP buffer was added to a final concentration of 10 nM, and the plate was incubated at room temperature for 30 min.
HBST2BPバッファーで同量のStv-HRPと複合体を形成したビオチン化検出モノボディ(9クローンとWT)を最終濃度10 nMになるように加え、プレートを室温で30分間インキュベートしました。
The microplate was washed five times with HBST2 buffer, and 50 μL of SuperSignal ELISA femto (Thermo Fisher Scientific) was added to each well.
マイクロプレートをHBST2バッファーで5回洗浄し、50μLのSuperSignal ELISAフェムト(Thermo Fisher Scientific)を各ウェルに加えました。
The chemiluminescence was immediately recorded using the SpectraMax M5 plate reader.
化学発光は、SpectraMax M5プレートリーダーを使用してすぐに記録されました。
For the other sandwich ELISA, a microplate with 96 wells was coated with 100 μL of 100 nM monobody clone 10 in HBS2 buffer overnight.
他のサンドイッチELISAの場合、96ウェルのマイクロプレートは、HBS2バッファー中の100 nLモノボディクローン10で100μLで一晩コーティングされました。
The microplate was washed once with HBST2, blocked with 1%(w/v) BSA in HBS2 buffer at room temperature for one hour, and was washed once with HBST2 buffer.
マイクロプレートをHBST2で1回洗浄し、HBS2バッファー中の1%(w / v)BSAで室温で1時間ブロックし、HBST2バッファーで1回洗浄しました。
One of the analytes, either the SARS-CoV-2 S1 subunit, the spike protein trimer, or the SARS-CoV S1 subunit was incubated with the detecting monobody–HRP (clone 12, 1 nM) on ice for one hour.
分析物の1つであるSARS-CoV-2 S1サブユニット、スパイクプロテイントリマー、またはSARS-CoV S1サブユニットのいずれかを、検出用モノボディHRP(クローン12、1 nM)と氷上で1時間インキュベートしました。
The solution was then added to the capturing monobody-immobilized microplate and was incubated at room temperature for one hour.
次に、この溶液を捕捉用モノボディ固定化マイクロプレートに加え、室温で1時間インキュベートしました。
The microplate was washed five times with HBST2 buffer and 200 μL of SuperSignal ELISA femto was added to each well.
マイクロプレートをHBST2バッファーで5回洗浄し、200μLのSuperSignal ELISAフェムトを各ウェルに添加しました。
The chemiluminescence was immediately recorded using the SpectraMax M5 plate reader.
化学発光は、SpectraMax M5プレートリーダーを使用して直ちに記録されました。
Binding assay of monobodies toward SARS-CoV2 particles
SARS-CoV2粒子に対するモノボディの結合アッセイ
The SARS-CoV-2 virus was isolated fromVero-E6 cells (ATCC) from a nasal swab sample of a patient in the Nagoya Medical Center, Japan.
SARS-CoV-2ウイルスは、日本の名古屋医療センターの患者の鼻腔スワブサンプルから、Vero-E6細胞(ATCC)から分離されました。
Virus-infected cells were grown in Dulbecco's modified Eagle medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum and penicillin (100 U/mL) and streptomycin (100 μg/mL) (Thermo Fisher Scientific).
ウイルスに感染した細胞は、10%ウシ胎児血清とペニシリン(100 U / mL)とストレプトマイシン(100μg/ mL)(Thermo Fisher Scientific)を添加したダルベッコの改変イーグル培地(Sigma-Aldrich)で培養しました。
Four days after infection, the supernatant was harvested.
感染から4日後、上澄みを回収しました。
The supernatant was clarified by centrifugation and filtration through a 0.45-μm-pore-size syringe filter (Merck Millipore), and used for the virion-binding assay.
上清を遠心分離し、孔径0.45μmのシリンジフィルター(Merck Millipore)で濾過して清澄化し、ビリオン結合アッセイに使用しました。
The virus particle amounts were measured using droplet digital PCR using the One Step RTddPCR Advanced Kit for Probes (Bio-Rad) with a primer/probe set for N2 (Data file S1).
ウイルス粒子の量は、N2用のプライマー/プローブセットを備えたOne Step RTddPCR Advanced Kits for Probes(Bio-Rad)を使用した液滴デジタルPCRを使用して測定しました(データファイルS1)。
SARS-CoV-2 particles (5×104 particles/140 μL) and 10 nM monobody in HBST buffer were incubated at 25°C for 10 min.
SAB-CoV-2粒子(5×104粒子/ 140μL)とHBSTバッファー中の10 nMモノボディを25℃で10分間インキュベートしました。
This was followed by addition of Dynabeads M-280 streptavidin (10 mg/mL, 10 μL, pre-washed by PBS buffer), which were incubated with the solution for 10 min.
その後、Dynabeads M-280ストレプトアビジン(10 mg / mL、10μL、PBSバッファーで事前洗浄)を追加し、溶液と10分間インキュベートしました。
After the incubation, the supernatant was collected for further analysis.
インキュベーション後、さらなる分析のために上清を収集した。
The beads were washed three times with HBST, and were suspended with 140 μL of HBST buffer.
ビーズをHBSTで3回洗浄し、140μLのHBSTバッファーで懸濁した。
Viral RNA was extracted from both the supernatant and the bead suspension using the QIAamp Viral RNA Mini Kit (Qiagen) as described in the manufacture′s protocol.
メーカーのプロトコルに記載されているように、QIAamp Viral RNA Mini Kit(Qiagen)を使用して、上清とビーズ懸濁液の両方からウイルスRNAを抽出しました。
The 5 μL out of the 60 μL RNAcontaining solution was added to a RT-qPCR reaction mixture (One Step PrimeScript III RT-qPCR Mix, TAKARA-Bio) using a primer/probe set for N2.
60μLのRNAを含む溶液から5μLを、N2用のプライマー/プローブセットを使用して、RT-qPCR反応混合物(One Step PrimeScript III RT-qPCR Mix、TAKARA-Bio)に添加しました。
The target copy numbers were determined using Thermal Cycler Dice Real Time System III (TAKARA-bio), which constantly showed about 3- times higher number of RNA genomes than that of above RTddPCR.
ターゲットコピー数は、Thermal Cycler Dice Real Time System III(TAKARA-bio)を使用して決定されました。これは、上記のRTddPCRよりも約3倍多い数のRNAゲノムを常に示しています。
Pull-down RT-qPCR for study of the detection limit of SARS-CoV2 particles
プルダウンRT-qPCRによる、SARS-CoV2粒子の検出限界の研究
SARS-CoV-2 virus particles (0.1–10,000 particles/μL based on viral RNA coy number, 700 μL) were incubated with 2 nM monobody clone 4 in PBS (10 mM phosphate pH 7.4, 2.7 mM KCl, 137 mM NaCl) containing 0.05%(v/v) Tween 20 at room temperature for 10 min.
SARS-CoV-2ウイルス粒子(ウイルスRNAのコイル数に基づいて0.1〜10,000粒子/μL、700μL)を、0.05%(v / v)Tween 20を含むPBS(10 mMリン酸pH 7.4、2.7 mM KCl、137 mM NaCl)内の2 nMモノボディクローン4と、室温で10分間インキュベートしました。
The solution was mixed with Dynabeads M-280 streptavidin (10 mg/mL, 8 μL, pre-washed by PBS buffer) at 25°C for 10 min.
溶液をDynabeads M-280ストレプトアビジン(10 mg / mL、8μL、PBSバッファーで事前洗浄)と25℃で10分間混合しました。
The beads were washed with HBST three times and were suspended in HBST.
ビーズをHBSTで3回洗浄し、HBSTに懸濁しました。
Viral RNA from the bead suspension or the original solution (14 μL) were extracted using the QIAamp Viral RNA Mini Kit.
QIAamp Viral RNA Mini Kitを使用して、ビーズ懸濁液からのウイルスRNAまたは元の溶液(14μL)を抽出しました。
For RT-qPCR, the One Step PrimeScript III RT-qPCR Mix with the N2 primer/probe set was used.
RT-qPCRには、N2プライマー/プローブセットを含むOne Step PrimeScript III RT-qPCR Mixを使用しました。
The target copy numbers were determined using Thermal Cycler Dice Real Time System III (TAKARA-bio) as aforementioned.
目標コピー数は、前述のようにThermal Cycler Dice Real Time System III(TAKARA-bio)を使用して決定されました。
Pull-down of SARS-CoV-2 in nasal swab specimens from patients
患者の鼻腔スワブ検体におけるSARS-CoV-2のプルダウン
For the pull-down assay using nasal swab specimens from patients, we used residual nasal samples after diagnostic tests.
患者の鼻腔スワブ検体を使用したプルダウンアッセイでは、診断テスト後に残った鼻腔サンプルを使用しました。
The study was approved by the ethical committee at the Nagoya Medical Center (registration #2019 -087) and conducted according to the tenets of the Declaration of Helsinki.
この研究は、名古屋医療センターの倫理委員会(登録番号2019-087)によって承認され、ヘルシンキ宣言の理念に従って実施されました。
※ヘルシンキ宣言=、世界医師会(WMA)によって作成された人体実験に関する一連の倫理的原則。ナチスによる人体実験への反省に基づく。
Written informed consent for use of the residual samples were obtained from all participants.
残りのサンプルの使用について、書面によるインフォームドコンセントがすべての参加者から取得されました。
35μL of each nasal sample was mixed with 2 nM monobody clone 4 to a total volume of 100 μL in HBST buffer and incubated at 25°C for 10 min.
各鼻腔サンプル35μLを2 nMモノボディクローン4と混合して、HBSTバッファーで総量100μLにし、25℃で10分間インキュベートしました。
The mixture was then incubated for another 10 min with Dynabeads M-280 streptavidin (2.5 mg/mL, 40 μL, pre-washed by HBST buffer).
次に、混合物をDynabeads M-280ストレプトアビジン(2.5 mg / mL、40μL、HBSTバッファーで前洗浄)とさらに10分間インキュベートしました。
The RNA copies in both the supernatant and the bead fraction were measured using RT-qPCR, as described above.
上清とビーズ画分の両方のRNAコピーは、上記のようにRT-qPCRを使用して測定されました。
ull-down direct RT-qPCR assay of SARS-CoV-2 in nasal swab specimens from patients
患者の鼻腔スワブ検体におけるSARS-CoV-2の完全ダウンダイレクトRT-qPCRアッセイ
Each residual sample of nasal swab from 4 patients was used for this assay.
このアッセイでは、4人の患者の鼻腔スワブの残りの各サンプルを使用しました。
Briefly, 0.5% Tween-20 (45 μL) and monobody clone 4 (5 μL of 200 nM) were added into 400 μL of the swab sample in PBS.
簡単に言えば、0.5%Tween-20(45μL)とモノボディクローン4(200 nMの5μL)をPBS内の400μLの綿棒サンプルに加えました。
The mixture was incubated at 25°C for 10 min, followed by addition of Dynabeads M-280 streptavidin (2.5 mg/mL, 10 μL, pre-washed by HBST buffer).
混合物を25°Cで10分間インキュベートした後、Dynabeads M-280ストレプトアビジン(2.5 mg / mL、10μL、HBSTバッファーで事前洗浄)を追加しました。
After 10-min incubation, the beads were washed with 1 mL of HBST three times and resuspended with 2.5 μL of HBST containing carrier RNA.
10分のインキュベーション後、ビーズを1 mLのHBSTで3回洗浄し、2.5μLのHBST含有キャリアRNAで再懸濁しました。
The bead suspension or the original swab solution (2.5 μL) were mixed with a RT-qPCR reaction mixture (47.5 μL, the PrimeDirect Probe RT-qPCR Mix, TAKARA-bio) with a primer/probe set (Data file S1).
ビーズ懸濁液または元の綿棒溶液(2.5μL)は、プライマー/プローブセット(データファイルS1)を含むRT-qPCR反応混合物(47.5μL、PrimeDirect Probe RT-qPCR Mix、TAKARA-bio)と混合されました。
The RNA copy numbers were determined using the Thermal Cycler Dice Real Time System III (TAKARA-bio) as aforementioned.
RNAコピー数は、前述のサーマルサイクラーダイスリアルタイムシステムIII(TAKARA-bio)を使用して決定されました。
The detection level of positive control RNA using the PrimeDirect Probe RTqPCR Mix was >~25 copies/well.
PrimeDirect Probe RTqPCR Mixを使用したポジティブコントロールRNAの検出レベルは、ウェルあたり25コピー以上でした。
Virus neutralization assay
ウイルス中和アッセイ
SARS-CoV-2 neutralization assay was performed using VeroE6/TMPRSS2 cells that were obtained from JCRB cell bank, Ibaraki, Japan.
SARS-CoV-2中和アッセイは、日本の茨城県のJCRBセルバンクから入手したVeroE6 / TMPRSS2細胞を使用して行われました。
The cells (5×103 cells per well, 50 μL) were seeded in 96-well culture plates and incubated at 37°C for 18 hours before infection.
細胞(ウェルあたり5×103細胞、50μL)を96ウェル培養プレートに播種し、感染前に37℃で18時間インキュベートしました。
Monobodies were four-fold serially diluted (from 400 nM to 24.4 pM) and 20 μL of the diluted monobodies were added into each culture well.
モノボディを4倍に順次希釈し(400 nMから24.4 pMまで)、20μLの希釈モノボディを各培養ウェルに加えました。
Cells were infected with 10 μL of SARS-CoV-2 (105 TCID50/mL) for 1 hour at 37°C.
37°Cで1時間、細胞に10μLのSARS-CoV-2(105 TCID50 / mL)を感染させました。
The supernatant was removed, and 80 μL of Dulbecco's modified Eagle medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum and penicillin (100 U/mL) and streptomycin (100 μg/mL) (Thermo Fisher Scientific).
上清を取り除き、80μLのダルベッコの改変イーグル培地(Sigma-Aldrich)を10%ウシ胎児血清およびペニシリン(100 U / mL)そしてストレプトマイシン(100μg/ mL)で補足しました(Thermo Fisher Scientific)。
After incubation at 37°C supplied with 5% CO2 for 36 hours, the culture supernatants were harvested.
5%CO2を供給した37℃で36時間インキュベートした後、培養上清を回収しました。
The SARSCoV-2 RNA amounts in the supernatants were measured by RT-qPCR using the SARS-CoV-2 Direct Detection RT-qPCR kit (Takara Bio).
上清中のSARSCoV-2 RNA量は、SARS-CoV-2直接検出RT-qPCRキット(Takara Bio)を使用したRT-qPCRによって測定されました。
The half maximal inhibitory concentrations (IC50) were determined using GraphPad Prism version 6.0.
最大抑制濃度の半分(IC50)は、GraphPad Prismバージョン6.0を使用して決定されました。
以下省略。
この記事が気に入ったらサポートをしてみませんか?